


The article below curated from the work of:
Gary B. O’Mealey, William L. Berry, and Scott M. Plafker
Sulforaphane is a phytochemical, a substance within the isothiocyanate group of organosulfur compounds, found in cruciferous vegetables, such as broccoli, cabbage, cauliflower, and Brussels sprouts. It can also be found in bok choy, kale, collards, mustard greens and watercress. Research studies have shown that sulforaphane can help prevent various types of cancer by activating the production of Nrf2, or nuclear factor erythroid 2-related factor, a transcription factor which regulates protective antioxidant mechanisms that control the cell’s response to oxidants. The purpose of the following article is to describe the function of sulforaphane.
Table of Contents
Abstract
The KEAP1-Nrf2-ARE antioxidant system is a principal means by which cells respond to oxidative and xenobiotic stresses. Sulforaphane (SFN), an electrophilic isothiocyanate derived from cruciferous vegetables, activates the KEAP1-Nrf2-ARE pathway and has become a molecule-of-interest in the treatment of diseases in which chronic oxidative stress plays a major etiological role. We demonstrate here that the mitochondria of cultured, human retinal pigment epithelial (RPE-1) cells treated with SFN undergo hyperfusion that is independent of both Nrf2 and its cytoplasmic inhibitor KEAP1. Mitochondrial fusion has been reported to be cytoprotective by inhibiting pore formation in mitochondria during apoptosis, and consistent with this, we show Nrf2-independent, cytoprotection of SFN-treated cells exposed to the apoptosis-inducer, staurosporine. Mechanistically, SFN mitigates the recruitment and/or retention of the soluble fission factor Drp1 to mitochondria and to peroxisomes but does not affect overall Drp1 abundance. These data demonstrate that the beneficial properties of SFN extend beyond the activation of the KEAP1-Nrf2-ARE system and warrant further interrogation given the current use of this agent in multiple clinical trials.
Keywords: Sulforaphane, Nrf2, Drp1, Mitochondria, Fission, Fusion, Apoptosis
Introduction
Sulforaphane is an Nrf2-Independent Inhibitor of Mitochondrial Fission
Sulforaphane (SFN) is an isothiocyanate compound derived in the diet most commonly from cruciferous vegetables [56]. It is generated in plants as a xenobiotic response to predation via vesicular release of the hydrolytic enzyme myrosinase from damaged cells; this enzyme converts glucosinolates to isothiocyantes [42]. Over the last two decades, SFN has been extensively characterized for its reported anticancer, antioxidant, and antimicrobial properties [57]. Much of this efficacy has been attributed to the capacity of SFN to modulate the KEAP1-Nrf2-antioxidant response element (ARE) signaling pathway, although additional activities of the compound have been identified, including the inhibition of histone deacetylase activity and cell cycle progression [29]. Nrf2 is the master antioxidant transcription factor and under conditions of homeostasis, its stability is suppressed through the action of the cytoplasmic Cullin3KEAP1 ubiquitin ligase complex [20]. Specifically, Nrf2 is recruited to the Cullin3KEAP1 ligase by binding to the dimeric substrate adaptor KEAP1 and is subsequently modified with polyUb chains that target the transcription factor for proteasome-mediated degradation. This constitutive turnover limits the half-life of Nrf2 in unstressed cells to ~15 min [30], [33], [46], [55]. In response to numerous types of stress, most notably oxidative stress, KEAP1, a cysteine-rich protein, acts as a redox sensor, and oxidative modification of critical cysteines, particularly C151, of KEAP1 dissociates Nrf2-KEAP1 from CUL3 thereby preventing Nrf2 degradation [8], [20], [55]. Notably, SFN, and possibly other Nrf2 activators, mimic oxidative stress by modifying C151 of KEAP1 e.g. [21]. Stabilization of Nrf2 allows for its translocation to the nucleus where it induces the expression of a battery of Phase II antioxidant and detoxification genes. Nrf2 binds to the antioxidant response promoter elements (ARE) of its cognate target genes through heterodimerization with small Maf proteins [19]. This system presents a dynamic and sensitive response to indirect antioxidants like SFN, free radicals generated by the mitochondria [16], or other physiologic sources of oxidative stress [41].
Mitochondria are dynamic, subcellular organelles that regulate a host of cellular functions ranging from ATP production and intracellular calcium buffering to redox regulation and apoptosis [13], [49]. Mitochondria also represent the principal source of reactive oxygen species (ROS) within the cell. Proper regulation of mitochondrial function is therefore necessary for optimizing ATP production to meet cellular needs while simultaneously minimizing the potentially harmful effects of excessive free radical production. A critical requirement for fine modulation of mitochondrial function is the capacity for mitochondria to function both independently as biochemical machines and as part of a vast, responsive network.
Mitochondrial network morphology and function are determined by a regulated balance between fission and fusion. Mitochondrial fission is required for daughter cell inheritance of mitochondria during cell division [28] as well as for the selective, autophagic degradation of depolarized or damaged mitochondria, termed mitophagy [1]. Conversely, fusion is required for complementation of mitochondrial genomes and sharing of electron transport chain components between neighboring mitochondria [54]. At the molecular level, mitochondrial fission and fusion are regulated by large, dynamin-like GTPases. Three enzymes primarily regulate fusion: Mitofusins 1 and 2 (Mfn1/2) are two-pass outer membrane proteins that mediate outer membrane fusion via heterotypic interactions between adjacent mitochondria [15], [25], [37], while OPA1 is an inner membrane protein that simultaneously ensures matrix connectivity by regulating the melding of inner membranes [5]. The GTPase activity of all three proteins is required for robust fusion [5], [18], and OPA1 is further regulated by complex proteolysis within the mitochondrial inner membrane by the proteases OMA1 [14], PARL [6], and YME1L [45]. Importantly, intact mitochondrial membrane potential is required for efficient fusion in order to suppress integration of damaged and healthy mitochondria [26].
Mitochondrial fission is primarily catalyzed by a cytosolic protein called Dynamin-related protein 1 (Drp1/DNM1L). Drp1 is recruited from the cytosol to prospective sites of fission on the mitochondrial outer membrane [43]. The major receptors for Drp1 on the outer membrane are mitochondrial fission factor (Mff) [32] and, to a lesser extent, Fission 1 (Fis1) [51]. Additionally, a decoy receptor, MIEF1/MiD51, was discovered that acts to further limit the activity of Drp1 protein at potential fission sites [58]. Once docked at the mitochondrial outer membrane, Drp1 oligomerizes into spiral-like structures around the body of the mitochondrion and then utilizes the energy derived from GTP hydrolysis to mediate the physical scission of the mitochondrial outer and inner membranes [17]. Endoplasmic reticulum-derived tubules act as an initial constrictor of mitochondria prior to Drp1 oligomerization, underscoring the revelation that non-constricted mitochondria are wider than the permissive circumference of a completed Drp1 spiral [12]. Actin dynamics are also important for the ER-mitochondria interactions that precede mitochondrial fission [24]. In addition to its role in mitochondrial fission, Drp1 catalyzes the fission of peroxisomes [40].
Drp1 is very similar to the well-characterized dynamin protein in that both proteins contain an N-terminal GTPase domain, a Middle domain that is critical for self-oligomerization, and a C-terminal GTPase effector domain [31]. Drp1 achieves selectivity for mitochondrial membranes through a combination of interactions with its receptor proteins Mff and Fis1 and also through its affinity for the mitochondria-specific phospholipid cardiolipin via the unique B-insert domain of Drp1 [2]. Drp1 typically exists as a homotetramer in the cytoplasm, and higher order assembly at mitochondrial fission sites is mediated by the Middle domain of Drp1 [3].
Given the implicit link between mitochondrial function and the KEAP1-Nrf2-ARE pathway, we investigated the effects of Nrf2 activation on mitochondrial structure and function. We demonstrate here that SFN induces mitochondrial hyperfusion that, unexpectedly, is independent of both Nrf2 and KEAP1. This effect of SFN is through an inhibition of Drp1 function. We further demonstrate that SFN confers resistance to apoptosis that is Nrf2-independent and mimics that observed in cells depleted of Drp1. These data collectively indicate that in addition to stabilizing and activating Nrf2, SFN modulates mitochondrial dynamics and preserves cellular fitness and survival.
Results
Sulforaphane Induces Nrf2/KEAP1-Independent Hyperfusion of Mitochondria
In the course of studying the effects of Nrf2 activation on mitochondrial network dynamics, we discovered that treatment of immortalized, human retinal pigment epithelial (RPE-1) cells with sulforaphane (SFN), a potent activator of Nrf2 signaling, induced a robust fusion of the mitochondrial network when compared with vehicle-treated control cells (Fig. 1A and B). The morphology of the mitochondria in these cells greatly resembled that of the mitochondria in cells depleted by siRNA of endogenous Drp1, the principal mitochondrial fission factor (Fig. 1A). This result raised the intriguing idea that mitochondrial fission and fusion status responds directly to Nrf2 levels in the cell. However, stimulation of cells with other Nrf2 stabilizers and activators such as the proteasome inhibitor MG132, the pro-oxidant tBHQ, or knockdown of the Nrf2 inhibitor KEAP1 did not induce mitochondrial fusion (Fig. 1A and B). Stabilization of Nrf2 by these manipulations was confirmed by western blotting for endogenous Nrf2 (Fig. 1C). Furthermore, expression of Nrf2 was dispensable for SFN-induced mitochondrial fusion, as knockdown of endogenous Nrf2 with siRNA failed to counter this phenotype (Fig. 1D–F). Because SFN stimulates the KEAP1-Nrf2-ARE pathway by covalently modifying cysteine residues of KEAP1 [21], we knocked down KEAP1 to address whether SFN-induced mitochondrial hyperfusion is stimulated through a KEAP1-dependent, but Nrf2 independent pathway. However, depletion of KEAP1 also failed to abrogate SFN-induced mitochondrial fusion (Fig. 1G–I). In fact, SFN reversed the pro-fission morphology induced by depletion of KEAP1 (Fig. 1G, panel b versus panel d). These results indicate that SFN treatment causes mitochondrial fusion independent of the canonical KEAP1-Nrf2-ARE pathway and led us to interrogate whether SFN directly affects components of the mitochondrial fission or fusion machinery.
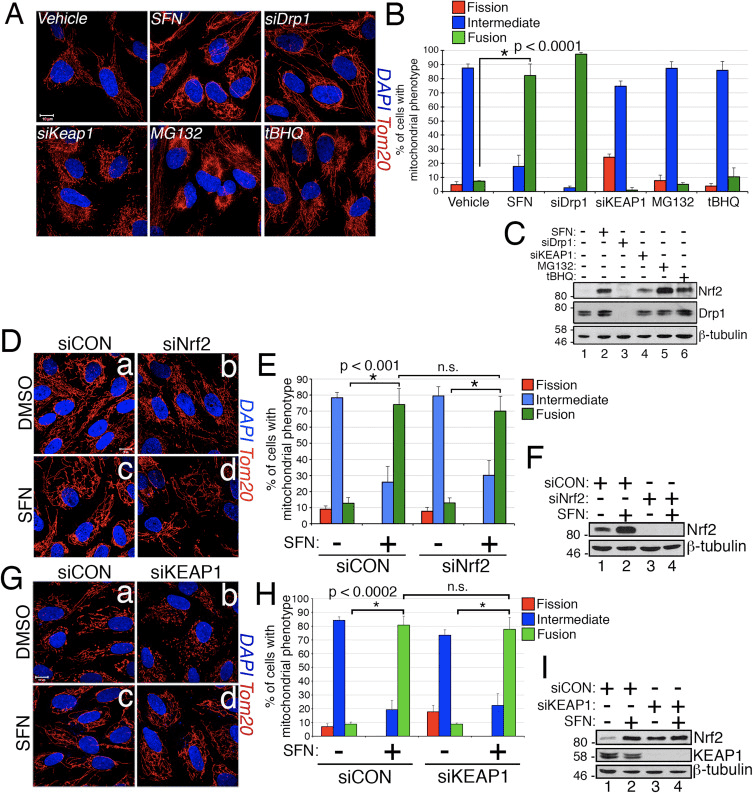
Sulforaphane Impairs the Mitochondrial Association of Drp1
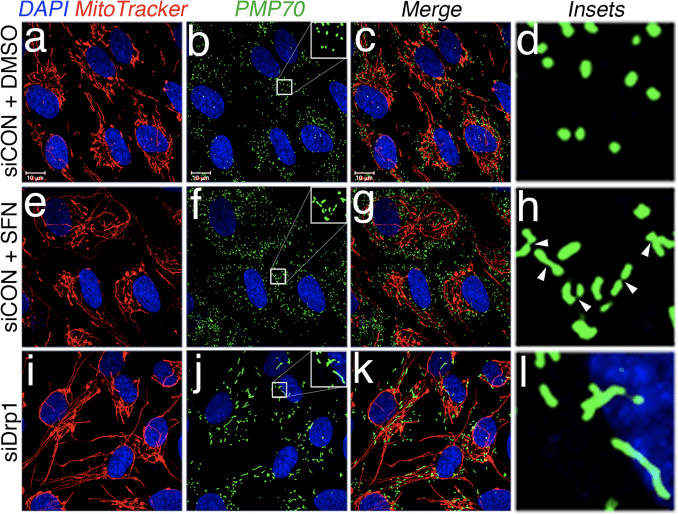
Based on the finding that SFN-treatment induces mitochondrial hyperfusion, we reasoned that this phenotype was either a consequence of excessive fusion activity or an inhibition of fission activity. To discriminate between these two possibilities, we compared the morphology of peroxisomes in the presence and absence of SFN. Peroxisomes are similar to mitochondria in that they are dynamic organelles the shape and length of which are constantly in flux [44]. Peroxisomes contain both Fis1 and Mff in their outer membrane and, as a consequence, are targets for Drp1-mediated fission [22], [23]. However, peroxisomes do not utilize the fusion machinery of the mitochondrial network and consequently, do not undergo fusion [39]. Rather, peroxisomal fission is opposed by the lengthening of existing peroxisomes via de novo addition of membranes and proteins [44]. Because peroxisomes lack Mfn1/2 and OPA1, we reasoned that if SFN activates the fusion machinery rather than inhibiting the fission machinery, peroxisome length would not be affected. In vehicle-treated cells, peroxisomes are maintained as short, round, punctiform organelles (Fig. 2, panels b and d). However, SFN treatment increased peroxisome length by ~2-fold as compared to control cells (Fig. 2, panels f and h). Furthermore, many of the peroxisomes were pinched near the center, indicating a potential scission defect (Fig. 2, panel h, arrowheads). Likewise, peroxisomes in cells transfected with Drp1 siRNA were abnormally long (Fig. 2, panels j and l), confirming that Drp1 is required for peroxisomal fission and suggesting that SFN-treatment causes mitochondrial and peroxisomal phenotypes by disrupting the fission machinery.
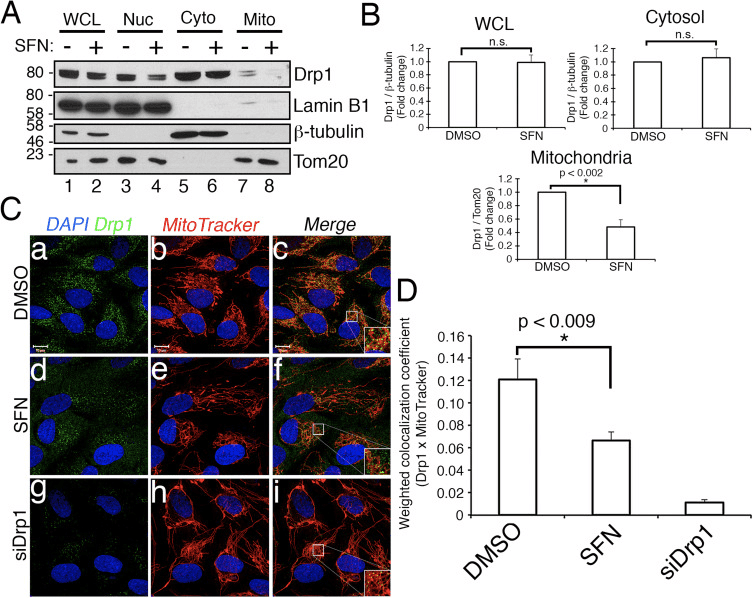
We next determined how SFN restricts Drp1 function. Possibilities included reductions in expression levels, recruitment/retention at mitochondria, oligomerization, or enzymatic activity of the GTPase. A deficit in any one of these would result in reduced mitochondrial fission and hyperfusion. We did not detect reproducible changes in Drp1 protein levels after SFN-treatment (Figs. 1C and 3A
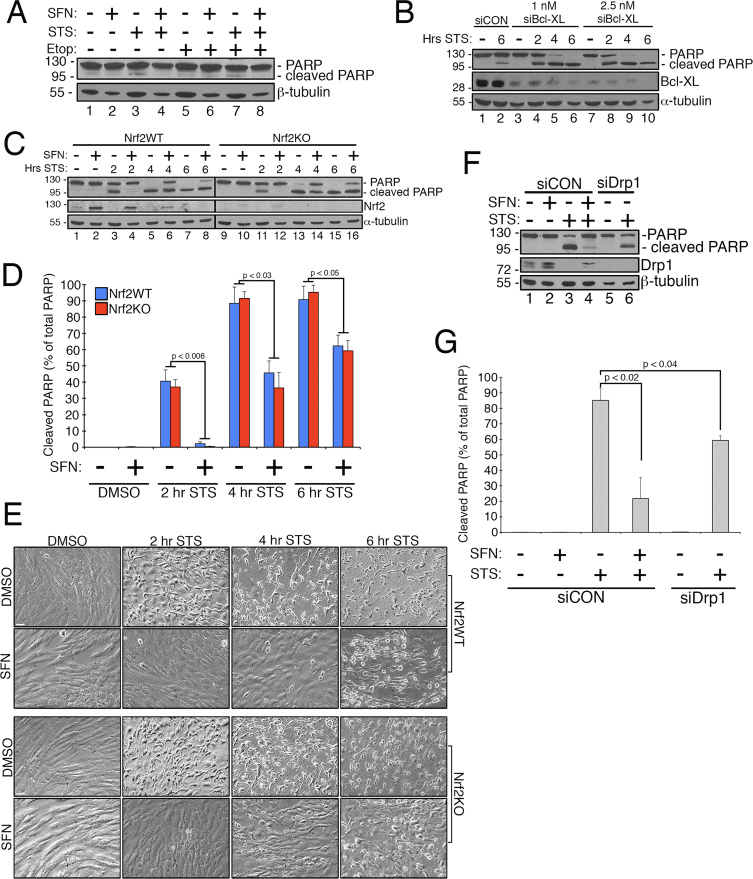
Sulforaphane Confers Protection Against Staurosportine-Induced Apoptosis Independent of Nrf2
Previous work has shown that mitochondrial fission is permissive in the formation of pores in the outer mitochondrial membrane generated by Bax/Bak during apoptosis [11]. Drp1 has been shown to be selectively recruited to mitochondria during apoptosis [11] and, consistent with this, fragmented mitochondria have been observed early in the process [27]. Conversely, inhibiting mitochondrial fission is thought to inhibit apoptosis by blocking the formation of the outer membrane pores that allow for cytochrome c release [53]. Accordingly, stimulating mitochondrial fusion delays the progression of apoptosis induced by compounds including staurosporine (STS) [14]. To determine whether SFN protects RPE-1 cells from STS-mediated apoptosis and if so, whether this requires Nrf2, we established an assay to readily induce poly ADP ribose polymerase (PARP) cleavage, a substrate of activated caspase-3 and
Discussion
We have discovered that SFN modulates mitochondrial fission/fusion dynamics independent of its effects on the KEAP1-Nrf2-ARE pathway. This is intriguing because of an assumed link between mitochondrial dysfunction and ROS production and the necessity of squelching mitochondria-derived free radicals through the activation of Nrf2. This additional functional impact of SFN is of potential importance given the more than 30 clinical trials currently underway testing SFN for the treatment of a variety of diseases including prostate cancer, obstructive pulmonary disease, and sickle cell disease [7], [10], [47].
Because SFN is an isothiocyanate [56] and it activates Nrf2 signaling by directly acylating critical KEAP1 cysteines to suppress Nrf2 degradation [21], it follows that SFN exerts its pro-fusion effects by modulating the activity of a fission or fusion factor via cysteine modification. Our data strongly support Drp1 being negatively regulated by SFN although whether the GTPase is a direct target of acylation remains to be elucidated. Despite this knowledge gap, the function of Drp1 is clearly being compromised by SFN as both mitochondria and peroxisomes become
Materials and Methods
Apoptosis Assays
Cells were seeded and transfected with siRNA as indicated below. The cells were pre-treated with 50 μM sulforaphane for 2 h to induce mitochondrial fusion and were then treated with 1 μM staurosporine to induce apoptosis. At the time of harvest, media was collected in individual tubes and subjected to high speed centrifugation to pellet apoptotic cells. This cell pellet was combined with adherent cells and solubilized in 2 times-concentrated Laemmli buffer. Samples were subjected to anti-PARP western blotting.
CRISPR/Cas9 Construct Generation
To create LentiCRISPR/eCas9 1.1, LentiCRISPR v2 (addgene #52961) was first cut with Age1 and BamH1. Next, SpCas9 from eSpCas9 1.1 (addgene #71814) was PCR amplified with Age1 and BamH1 overhangs using the following primers (Forward AGCGCACCGGTTCTAGAGCGCTGCCACCATGGACTATAAGGACCACGAC, Reverse AAGCGCGGATCCCTTTTTCTTTTTTGCCTGGCCGG) and ligated into the cut vector above. sgRNA sequences were determined by using Benchling.com. Parameters were set to target the coding sequence with the highest on-target and lowest off-target scores. The following sequences (targeting sequence underlined, hs sgNFE2L2#1 sense CACCGCGACGGAAAGAGTATGAGC, antisense AAACGCTCATACTCTTTCCGTCGC; hs sgNFE2L2#2 sense CACCGGTTTCTGACTGGATGTGCT, antisense AAACAGCACATCCAGTCAGAAACC; hs sgNFE2L2#3 sense CACCGGAGTAGTTGGCAGATCCAC, antisense AAACGTGGATCTGCCAACTACTCC) were annealed and ligated into BsmB1 cut LentiCRISPR/eCas9 1.1. Lentivirally infected RPE-1 cells were selected with puromycin and maintained as a pooled population. Knockout was confirmed by immunofluorescence and western blotting.
Cell Culture and Transfections
Human retinal pigment epithelial cells transformed with telomerase (RPE-1) (ATCC) were cultured in Dulbecco’s Modified Eagle Medium (DMEM) containing 1 g/L glucose supplemented with penicillin, streptomycin, 1X non-essential amino acid cocktail (Life Technologies), and 10% Fetal Bovine Serum (Life Technologies). For siRNA-transfections, 30,000–35,000 cells/mL were seeded overnight. Cells received 10 nM siRNA diluted in serum-free DMEM and combined with 0.3% Interferin transfection reagent (PolyPlus). For apoptosis sensitization, cells received 1 nM Bcl-XL siRNA. Cells were harvested 2–3 days post-transfection.
Chemicals, Antibodies, and siRNA Oligos
Antibodies against α-tubulin (Cell Signaling), β-tubulin (Sigma), Drp1 (BD Biosciences), KEAP1 (Proteintech), Lamin B1 (Abcam), PARP (Cell Signaling), PMP70 (Abcam), and Tom20 (BD Biosciences) were used at 1:1000 dilutions for western blotting and for immunofluorescence. In-house, anti-Nrf2 rabbit antibody was used at 1:2000 for western blotting [34], [59]. Sulforaphane (Sigma) and staurosporine (Tocris) were used at 50 μM and 1 μM respectively. siRNAs against Drp1 (Dharmacon), Nrf2 (Dharmacon), KEAP1 (Cell Signaling), and Bcl-XL (Cell Signaling) were used at 10 nM unless otherwise noted.
Immunofluorescence and in Vivo Labeling
Cells seeded on 18 mm glass coverslips were treated with vehicle or drug, fixed in 3.7% formaldehyde and then permeabilized in 0.2% Triton X-100/PBS on ice for 10 min. Primary antibodies were incubated in 3% bovine serum albumin (BSA) in PBS overnight at 4 °C. Following PBS washes, cells were incubated for 1 h in species-appropriate, Alexa488- or Alexa546-, conjugated secondary antibodies (diluted 1:1000) and 0.1 μg/mL DAPI (Sigma) in 3% BSA/PBS. Mitochondria were visualized either by anti-Tom20 immunofluorescence or by incubating cells in 200 nM MitoTracker Red CMXRos (Molecular Probes, Inc.) in serum-free DMEM for 30 min at 37 °C prior to fixation.
Microscopy and Image Analysis
Immunofluorescence samples were viewed on an LSM710 Confocal microscope (Carl Zeiss). Micrographs were captured using 63X or 100X oil immersion objectives and images adjusted and enhanced using Adobe Photoshop CS6. Co-localization analysis was performed using Carl Zeiss LSM710 co-localization feature with thresholds manually set while blinded to the identity of the samples. Scale bars throughout, unless otherwise indicated, are 10 µm. Mitochondrial morphology was assessed by blinded scoring. If the mitochondria of a cell were maintained as multiple, round, discriminate puncta, the cell was scored as ‘fission’. If individual mitochondria were indistinguishable and the whole mitochondrial network appeared continuous, the cell was scored as ‘fusion’. All other cells, including those with clustering mitochondria, were scored as ‘intermediate’.
Subcellular Fractionations
RPE-1 cells were grown to confluence. Following a PBS wash, cells were subjected to centrifugation at 600×g for 10 min and resuspended in 600 μL isolation buffer (210 mM Mannitol, 70 mM Sucrose, 5 mM MOPS, 1 mM EDTA pH 7.4+1 mM PMSF). The suspension was lysed 30 times in a Dounce homogenizer. A fraction of the homogenate was preserved as a “whole cell lysate.” The remainder was subjected to centrifugation at 800×g for 10 min to pellet nuclei. Supernatants were subjected to centrifugation at 1500×g for 10 min to clear remaining nuclei and unlysed cells. This supernatant was subjected to centrifugation at 15,000×g for 15 min to pellet mitochondria. The supernatant was preserved as the “cytosolic fraction”. The pellet was washed gently with PBS and resuspended in isolation buffer. The protein concentration of each fraction was measured by bicinchoninic acid (BCA) assay and equivalent amounts of protein were resolved by SDS-PAGE.
Western Blotting
Cells were washed in PBS and solubilized in 2 times concentrated Laemmli solubilizing buffer (100 mM Tris [pH 6.8], 2% SDS, 0.008% bromophenol blue, 2% 2-mercaptoethanol, 26.3% glycerol, and 0.001%

Sulforaphane is a chemical from the isothiocyanate collection of organosulfur substances obtained from cruciferous vegetables, including broccoli, cabbage, cauliflower, kale, and collards, among others. Sulforaphane is produced when the enzyme myrosinase transforms glucoraphanin, a glucosinolate, into sulforaphane, also known as sulforaphane-glucosinolate. Broccoli sprouts and cauliflower have the highest concentration of glucoraphanin or the precursor to sulforaphane. Research studies have demonstrated that sulforaphane enhances the human body’s antioxidant capabilities to prevent various health issues.
Dr. Alex Jimenez D.C., C.C.S.T. Insight
Sulforaphane and Its Effects on Cancer, Mortality, Aging, Brain and Behavior, Heart Disease & More
Isothiocyanates are some of the most important plant compounds you can get in your diet. In this
Key sections:
- 00:01:14 – Cancer and mortality
- 00:19:04 – Aging
- 00:26:30 – Brain and behavior
- 00:38:06 – Final recap
- 00:40:27 – Dose
Full timeline:
- 00:00:34 – Introduction of sulforaphane, a major focus of the video.
- 00:01:14 – Cruciferous vegetable consumption and reductions in all-cause mortality.
- 00:02:12 – Prostate cancer risk.
- 00:02:23 – Bladder cancer risk.
- 00:02:34 – Lung cancer in smokers risk.
- 00:02:48 – Breast cancer risk.
- 00:03:13 – Hypothetical: what if you already have cancer? (interventional)
- 00:03:35 – Plausible mechanism driving
the cancer and mortality associative data. - 00:04:38 – Sulforaphane and cancer.
- 00:05:32 – Animal evidence showing
strong effect of broccoli sprout extract on bladder tumor development in rats. - 00:06:06 – Effect of direct supplementation of sulforaphane in prostate cancer patients.
- 00:07:09 – Bioaccumulation of isothiocyanate metabolites in actual breast tissue.
- 00:08:32 – Inhibition of breast cancer stem cells.
- 00:08:53 – History lesson: brassicas were established as having health properties even in ancient Rome.
- 00:09:16 – Sulforaphane’s ability to enhance carcinogen excretion (benzene, acrolein).
- 00:09:51 – NRF2 as a genetic switch via antioxidant response elements.
- 00:10:10 – How NRF2 activation enhances carcinogen excretion via glutathione-S-conjugates.
- 00:10:34 – Brussels sprouts increase glutathione-S-transferase and reduce DNA damage.
- 00:11:20 – Broccoli sprout drink increases benzene excretion by 61%.
- 00:13:31 – Broccoli sprout homogenate increases antioxidant enzymes in the upper airway.
- 00:15:45 – Cruciferous vegetable consumption and heart disease mortality.
- 00:16:55 – Broccoli sprout powder improves blood lipids and overall heart disease risk in type 2 diabetics.
- 00:19:04 – Beginning of
aging section. - 00:19:21 – Sulforaphane-enriched diet enhances
lifespan of beetles from 15 to 30% (in certain conditions). - 00:20:34 – Importance of low inflammation for longevity.
- 00:22:05 – Cruciferous vegetables and broccoli sprout powder seem to reduce a wide variety of inflammatory markers in humans.
- 00:23:40 – Mid-video recap: cancer, aging sections
- 00:24:14 – Mouse studies suggest sulforaphane might improve adaptive immune function in old age.
- 00:25:18 – Sulforaphane improved hair growth in a mouse model of balding.
Picture at 00:26:10. - 00:26:30 – Beginning of brain and behavior section.
- 00:27:18 – Effect of broccoli sprout extract on autism.
- 00:27:48 – Effect of glucoraphanin on schizophrenia.
- 00:28:17 – Start of depression discussion (plausible mechanism and studies).
- 00:31:21 – Mouse study using 10 different models of stress-induced depression show sulforaphane similarly effective as fluoxetine (
prozac ). - 00:32:00 – Study shows direct ingestion of glucoraphanin in mice is similarly effective at preventing depression from social defeat stress model.
- 00:33:01 – Beginning of neurodegeneration section.
- 00:33:30 – Sulforaphane and Alzheimer’s disease.
- 00:33:44 – Sulforaphane and Parkinson’s disease.
- 00:33:51 – Sulforaphane and Hungtington’s disease.
- 00:34:13 – Sulforaphane increases heat shock proteins.
- 00:34:43 – Beginning of traumatic brain injury section.
- 00:35:01 – Sulforaphane injected immediately after TBI improves memory (mouse study).
- 00:35:55 – Sulforaphane and neuronal plasticity.
- 00:36:32 – Sulforaphane improves learning in
model of type II diabetes in mice. - 00:37:19 – Sulforaphane and
duchenne muscular dystrophy. - 00:37:44 – Myostatin inhibition in muscle satellite cells (in vitro).
- 00:38:06 – Late-video recap: mortality and cancer, DNA damage, oxidative stress and inflammation, benzene excretion, cardiovascular disease, type II diabetes, effects on the brain (depression, autism, schizophrenia, neurodegeneration), NRF2 pathway.
- 00:40:27 – Thoughts on figuring out a dose of broccoli sprouts or sulforaphane.
- 00:41:01 – Anecdotes on sprouting at home.
- 00:43:14 – On cooking temperatures and sulforaphane activity.
- 00:43:45 – Gut bacteria conversion of sulforaphane from glucoraphanin.
- 00:44:24 – Supplements work better when combined with active myrosinase from vegetables.
- 00:44:56 – Cooking techniques and cruciferous vegetables.
- 00:46:06 – Isothiocyanates as goitrogens.
Acknowledgements
Sciencedirect.com/science/article/pii/S2213231716302750
How is Sulforaphane Produced?
Heating Decreases Epithiospecifier Protein Activity and Increases Sulforaphane Formation in Broccoli
Abstract
Sulforaphane, an isothiocyanate from broccoli, is one of the most potent food-derived anticarcinogens. This compound is not present in the intact vegetable, rather it is formed from its glucosinolate precursor, glucoraphanin, by the action of myrosinase, a thioglucosidase enzyme, when broccoli tissue is crushed or chewed. However, a number of studies have demonstrated that sulforaphane yield from glucoraphanin is

Pre-heating broccoli florets and sprouts to 60 °C significantly increased the myrosinase-catalyzed formation of sulforaphane (SF) in vegetable tissue extracts after crushing. This was associated with decreases in sulforaphane nitrile (SF Nitrile) formation and epithiospecifier protein (ESP) activity.
Keywords: Broccoli, Brassica oleracea, Cruciferae, Cancer, Anticarcinogen, Sulforaphane, Sulforaphane nitrile, Epithiospecifier protein, Quinone reductase
In conclusion, sulforaphane is a phytochemical found in
Curated by Dr. Alex Jimenez
Referenced from: Sciencedirect.com

Additional Topic Discussion: Acute Back Pain
Back pain is one of the most prevalent causes of disability and missed days at work worldwide. Back pain attributes to the second most common reason for doctor office visits, outnumbered only by upper-respiratory infections. Approximately 80 percent of the population will experience back pain at least once throughout their life. The spine is a complex structure made up of bones, joints, ligaments, and muscles, among other soft tissues. Because of this, injuries and/or aggravated conditions, such as herniated discs, can eventually lead to symptoms of back pain. Sports injuries or automobile accident injuries are often the most frequent cause of back pain, however, sometimes the simplest of movements can have painful results. Fortunately, alternative treatment options, such as chiropractic care, can help ease back pain through the use of spinal adjustments and manual manipulations, ultimately improving pain relief.

EXTRA EXTRA | IMPORTANT TOPIC: Recommended El Paso, TX Chiropractor
***
Post Disclaimer
Professional Scope of Practice *
The information on this blog site is not intended to replace a one-on-one relationship with a qualified healthcare professional or licensed physician and is not medical advice. We encourage you to make healthcare decisions based on your research and partnership with a qualified healthcare professional.
Blog Information & Scope Discussions
Welcome to El Paso's Premier Wellness and Injury Care Clinic & Wellness Blog, where Dr. Alex Jimenez, DC, FNP-C, a board-certified Family Practice Nurse Practitioner (FNP-BC) and Chiropractor (DC), presents insights on how our team is dedicated to holistic healing and personalized care. Our practice aligns with evidence-based treatment protocols inspired by integrative medicine principles, similar to those found on this site and our family practice-based chiromed.com site, focusing on restoring health naturally for patients of all ages.
Our areas of chiropractic practice include Wellness & Nutrition, Chronic Pain, Personal Injury, Auto Accident Care, Work Injuries, Back Injury, Low Back Pain, Neck Pain, Migraine Headaches, Sports Injuries, Severe Sciatica, Scoliosis, Complex Herniated Discs, Fibromyalgia, Chronic Pain, Complex Injuries, Stress Management, Functional Medicine Treatments, and in-scope care protocols.
Our information scope is limited to chiropractic, musculoskeletal, physical medicine, wellness, contributing etiological viscerosomatic disturbances within clinical presentations, associated somato-visceral reflex clinical dynamics, subluxation complexes, sensitive health issues, and functional medicine articles, topics, and discussions.
We provide and present clinical collaboration with specialists from various disciplines. Each specialist is governed by their professional scope of practice and their jurisdiction of licensure. We use functional health & wellness protocols to treat and support care for the injuries or disorders of the musculoskeletal system.
Our videos, posts, topics, subjects, and insights cover clinical matters and issues that relate to and directly or indirectly support our clinical scope of practice.*
Our office has made a reasonable effort to provide supportive citations and has identified relevant research studies that support our posts. We provide copies of supporting research studies available to regulatory boards and the public upon request.
We understand that we cover matters that require an additional explanation of how they may assist in a particular care plan or treatment protocol; therefore, to discuss the subject matter above further, please feel free to ask Dr. Alex Jimenez, DC, APRN, FNP-BC, or contact us at 915-850-0900.
We are here to help you and your family.
Blessings
Dr. Alex Jimenez DC, MSACP, APRN, FNP-BC*, CCST, IFMCP, CFMP, ATN
email: coach@elpasofunctionalmedicine.com
Licensed as a Doctor of Chiropractic (DC) in Texas & New Mexico*
Texas DC License # TX5807
New Mexico DC License # NM-DC2182
Licensed as a Registered Nurse (RN*) in Texas & Multistate
Texas RN License # 1191402
ANCC FNP-BC: Board Certified Nurse Practitioner*
Compact Status: Multi-State License: Authorized to Practice in 40 States*
Graduate with Honors: ICHS: MSN-FNP (Family Nurse Practitioner Program)
Degree Granted. Master's in Family Practice MSN Diploma (Cum Laude)
Dr. Alex Jimenez, DC, APRN, FNP-BC*, CFMP, IFMCP, ATN, CCST
My Digital Business Card