


Biochemistry of Pain: All pain syndromes have an inflammation profile. An inflammatory profile can vary from person to person and can also vary in one person at different times. The treatment of pain syndromes is to understand this inflammation profile. Pain syndromes are treated medically, surgically or both. The goal is to inhibit/suppress the production of inflammatory mediators. And a successful outcome is one that results in less inflammation and of course less pain.
Table of Contents
Biochemistry Of Pain
Objectives:
- Who are the key players
- What are the biochemical mechanisms?
- What are the consequences?
Inflammation Review:
Key Players
Why Does My Shoulder Hurt? A Review Of The Neuroanatomical & Biochemical Basis Of Shoulder Pain
ABSTRACT
If a patient asks ‘why does my shoulder hurt?’ the conversation will quickly turn to scientific theory and sometimes unsubstantiated conjecture. Frequently, the clinician becomes aware of the limits of the scientific basis of their explanation, demonstrating the incompleteness of our understanding of the nature of shoulder pain. This review takes a systematic approach to help answer fundamental questions relating to shoulder pain, with a view to providing insights into future research and novel methods for treating shoulder pain. We shall explore the roles of (1) the peripheral receptors, (2) peripheral pain processing or ‘nociception’, (3) the spinal cord, (4) the brain, (5) the location of receptors in the shoulder and (6) the neural anatomy of the shoulder. We also consider how these factors might contribute to the variability in the clinical presentation, the diagnosis and the treatment of shoulder pain. In this way we aim to provide an overview of the component parts of the peripheral pain detection system and central pain processing mechanisms in shoulder pain that interact to produce clinical pain.
INTRODUCTION: A VERY BRIEF HISTORY OF PAIN SCIENCE ESSENTIAL FOR CLINICIANS
The nature of pain, in general, has been a subject of much controversy over the past century. In the 17th century Descartes’ theory1 proposed that the intensity of pain was directly related to the amount of associated tissue injury and that pain was processed in one distinct pathway. Many earlier theories relied upon this so-called ‘dualist’ Descartian philosophy, seeing pain as the consequence of the stimulation of a ‘specific’ peripheral pain receptor in the brain. In the 20th century a scientific battle between two opposing theories ensued, namely specificity theory and pattern theory. The Descartian ‘specificity theory’ saw pain as a specific separate modality of sensory input with its own apparatus, while ‘pattern theory’ felt that pain resulted from the intense stimulation of non-specific receptors.2 In 1965, Wall and Melzack’s 3 gate theory of pain provided evidence for a model in which pain perception was modulated by both sensory feedback and the central nervous system. Another huge advance in pain theory at around the same time saw the discovery of the specific mode of actions of the opioids.4 Subsequently, recent advances in neuroimaging and molecular medicine have vastly expanded our overall understanding of pain.
So how does this relate to shoulder pain? Shoulder pain is a common clinical problem, and a robust understanding of the way in which pain is processed by the body is essential to best diagnose and treat a patient’s pain. Advances in our knowledge of pain processing promise to explain the mismatch between pathology and the perception of pain, they may also help us explain why certain patients fail to respond to certain treatments.
BASIC BUILDING BLOCKS OF PAIN
Peripheral sensory receptors: the mechanoreceptor and the ‘nociceptor’
There are numerous types of peripheral sensory receptors present in the human musculoskeletal system. 5 They may be classified based on their function (as mechanoreceptors, thermoreceptors or nociceptors) or morphology (free nerve endings or different types of encapsulated receptors).5 The different types of receptor can then be further subclassified based on the presence of certain chemical markers. There are significant overlaps between different functional classes of receptor, for example
Peripheral Pain Processing: ‘Nociception’
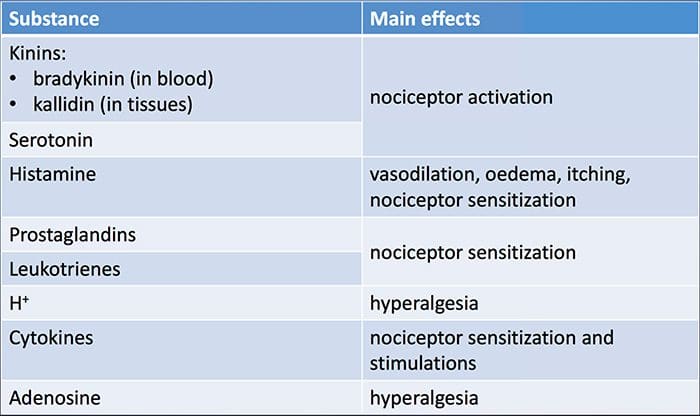
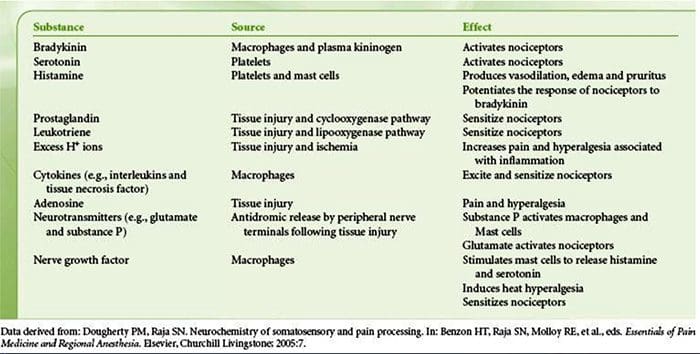
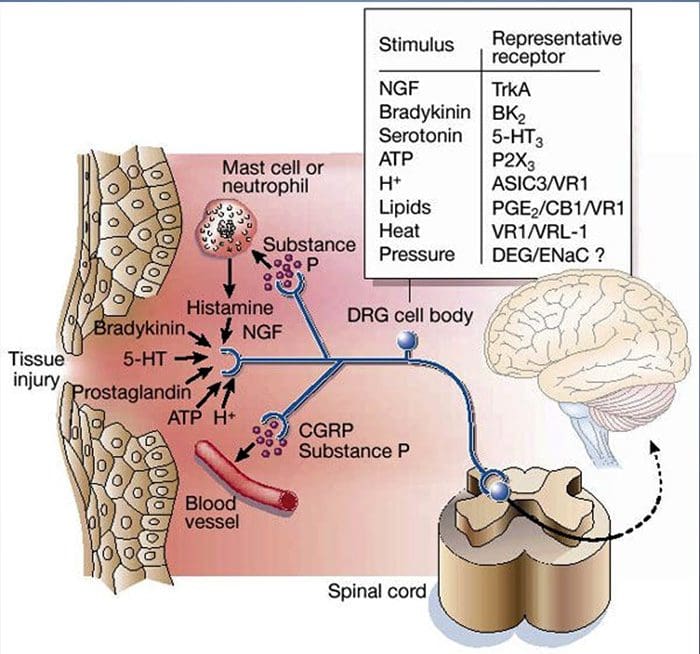
Tissue injury involves a variety of inflammatory mediators being released by damaged cells including bradykinin, histamine, 5-hydroxytryptamine, ATP, nitric oxide and certain ions (K+ and H+). The activation of the arachidonic acid pathway leads to the production of prostaglandins, thromboxanes and leuko- trienes. Cytokines, including the interleukins and tumor necrosis factor α, and neurotrophins, such as nerve growth factor (NGF), are also released and are intimately involved in the facilitation of inflammation.15 Other substances such as excitatory amino acids (glutamate) and opioids (endothelin-1) have also been implicated in the acute inflammatory response.16 17 Some of these agents may directly activate nociceptors, while others bring about the recruitment of other cells which then release further facilitatory agents.18 This local process resulting in the increased responsiveness of nociceptive neurons to their normal input and/or the recruitment of a response to normally subthreshold inputs is termed ‘peripheral sensitization’. Figure 1 summarizes some of the key mechanisms involved.
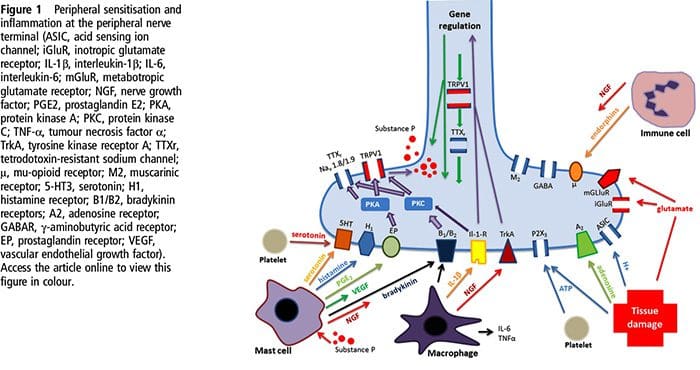
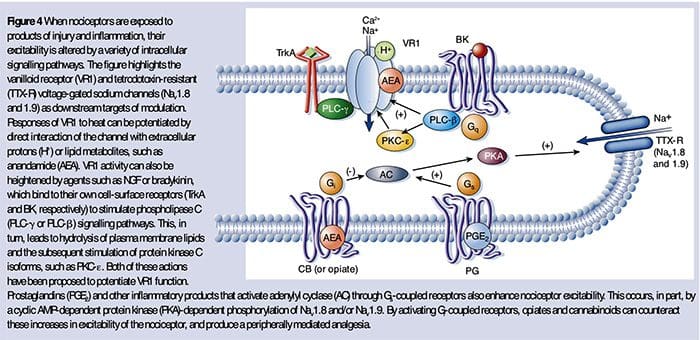
NGF and the transient receptor potential cation channel subfamily V member 1 (TRPV1) receptor have a symbiotic relationship when it comes to inflammation and nociceptor sensitization. The cytokines produced in inflamed tissue result in an increase in NGF production.19 NGF stimulates the release of histamine and serotonin (5-HT3) by mast cells, and also sensitizes nociceptors, possibly altering the properties of Aδ fibers such that a greater proportion become nociceptive. The TRPV1 receptor is present in a subpopulation of primary afferent fibers and is activated by capsaicin, heat and protons. The TRPV1 receptor is synthesized in the cell body of the afferent fibre, and is transported to both the peripheral and central terminals, where it contributes to the sensitivity of nociceptive afferents. Inflammation results in NGF production peripherally which then binds to the tyrosine kinase receptor type 1 receptor on the nociceptor terminals, NGF is then transported to the cell body where it leads to an up regulation of TRPV1 transcription and consequently increased nociceptor sensitivity.19 20 NGF and other inflammatory mediators also sensitize TRPV1 through a diverse array of secondary messenger pathways. Many other receptors including cholinergic receptors, γ-aminobutyric acid (GABA) receptors and somatostatin receptors are also thought to be involved in peripheral nociceptor sensitivity.
A large number of inflammatory mediators have been specifically implicated in shoulder pain and rotator cuff disease.21–25 While some chemical mediators directly activate nociceptors, most lead to changes in the sensory neuron itself rather than directly activating it. These changes may be early post- translational or delayed transcription dependent. Examples of the former are changes in the TRPV1 receptor or in voltage- gated ion channels resulting from the phosphorylation of membrane-bound proteins. Examples of the latter include the NGF-induced increase in TRV1 channel production and the calcium-induced activation of intracellular transcription factors.
Molecular Mechanisms Of Nociception
The sensation of pain alerts us to real or impending injury and triggers appropriate protective responses. Unfortunately, pain often outlives its usefulness as a warning system and instead becomes chronic and debilitating. This transition to a chronic phase involves changes within the spinal cord and brain, but there is also remarkable modulation where pain messages are initiated — at the level of the primary sensory neuron. Efforts to determine how these neurons detect pain-producing stimuli of a thermal, mechanical or chemical nature have revealed new signaling mechanisms and brought us closer to understanding the molecular events that facilitate transitions from acute to persistent pain.
The Neurochemistry Of Nociceptors
Glutamate is the predominant excitatory neurotransmitter in all nociceptors. Histochemical studies of adult DRG, however, reveal two broad classes of unmyelinated C fiber.
Chemical Transducers To Make The Pain Worse
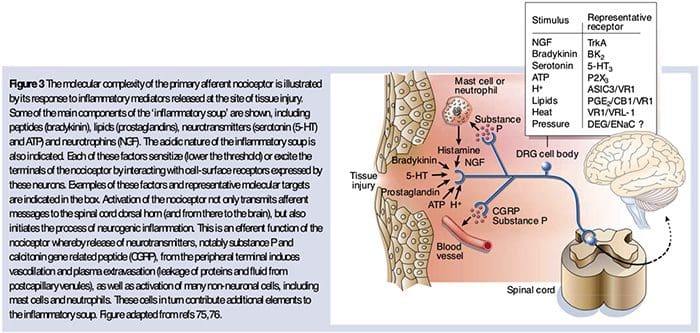
As described above, injury heightens our pain experience by increasing the sensitivity of nociceptors to both thermal and mechanical stimuli. This phenomenon results, in part, from the production and release of chemical mediators from the primary sensory terminal and from non-neural cells (for example, fibroblasts, mast cells, neutrophils and platelets) in the environment36 (Fig. 3). Some components of the inflammatory soup (for example, protons, ATP, serotonin or lipids) can alter neuronal excitability directly by inter- acting with ion channels on the nociceptor surface, whereas others (for example, bradykinin and NGF) bind to metabotropic receptors and mediate their effects through second-messenger signaling cascades11. Considerable progress has been made in understanding the biochemistry basis of such modulatory mechanisms.
Extracellular Protons & Tissue Acidosis
Local tissue acidosis is a hallmark physiological response to injury, and the degree of associated pain or discomfort is well correlated with the magnitude of acidification37. Application of acid (pH 5) to the skin produces sustained discharges in a third or more of polymodal nociceptors that innervate the receptive field 20.
Cellular & Molecular Mechanisms Of Pain
Abstract
The nervous system detects and interprets a wide range of thermal and mechanical stimuli as well as environmental and endogenous chemical irritants. When intense, these stimuli generate acute pain, and in the setting of persistent injury, both peripheral and central nervous system components of the pain transmission pathway exhibit tremendous plasticity, enhancing pain signals and producing hypersensitivity. When plasticity facilitates protective reflexes, it can be beneficial, but when the changes persist, a chronic pain condition may result. Genetic, electrophysiological, and pharmacological studies are elucidating the molecular mechanisms that underlie detection, coding, and modulation of noxious stimuli that generate pain.
Introduction: Acute Versus Persistent Pain
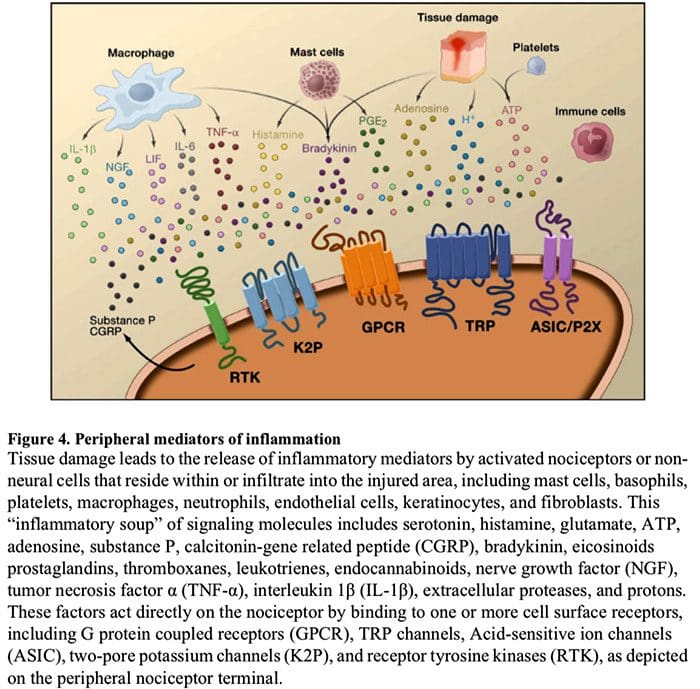
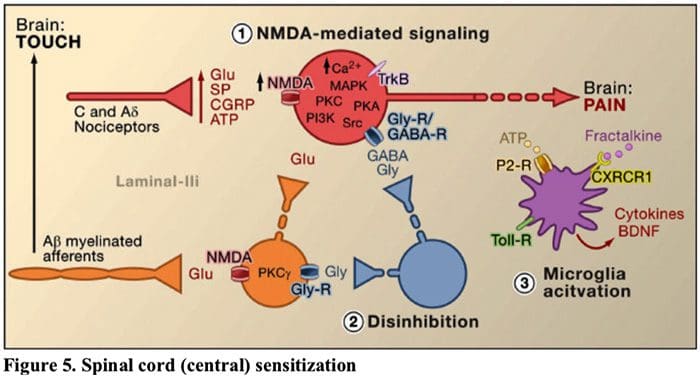
Figure 5. Spinal Cord (Central) Sensitization
- Glutamate/NMDA receptor-mediated sensitization. Following intense stimulation or persistent injury, activated C and Aδ nociceptors release a variety of neurotransmitters including dlutamate, substance P, calcitonin-gene related peptide (CGRP), and ATP, onto output neurons in lamina I of the superficial dorsal horn (red). As a consequence, normally silent NMDA glutamate receptors located in the postsynaptic neuron can now signal, increase intracellular calcium, and activate a host of calcium dependent signaling pathways and second messengers including mitogen-activated protein kinase (MAPK), protein kinase C (PKC), protein kinase A (PKA) and Src. This cascade of events will increase the excitability of the output neuron and facilitate the transmission of pain messages to the brain.
- Disinhibition. Under normal circumstances, inhibitory interneurons (blue) continuously release GABA and/or glycine (Gly) to decrease the excitability of lamina I output neurons and modulate pain transmission (inhibitory tone). However, in the setting of injury, this inhibition can be lost, resulting in hyperalgesia. Additionally, disinhibition can enable non-nociceptive myelinated Aβ primary afferents to engage the pain transmission circuitry such that normally innocuous stimuli are now perceived as painful. This occurs, in part, through the disinhibition of excitatory PKCγ expressing interneurons in inner lamina II.
- Microglial activation. Peripheral nerve injury promotes release of ATP and the chemokine fractalkine that will stimulate microglial cells. In particular, activation of purinergic, CX3CR1, and Toll-like receptors on microglia (purple) results in the release of brain-derived neurotrophic factor (BDNF), which through activation of TrkB receptors expressed by lamina I output neurons, promotes increased excitability and enhanced pain in response to both noxious and innocuous stimulation (that is, hyperalgesia and allodynia). Activated microglia also release a host of cytokines, such as tumor necrosis factor α (TNFα), interleukin-1β and 6 (IL-1β, IL-6), and other factors that contribute to central sensitization.
The Chemical Milieu Of Inflammation
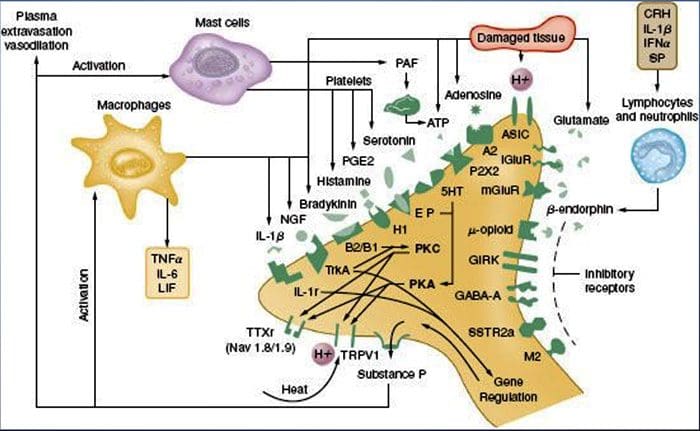
Peripheral sensitization more commonly results from inflammation-associated changes in the chemical environment of the nerve fiber (McMahon et al., 2008). Thus, tissue damage is often accompanied by the accumulation of endogenous factors released from activated nociceptors or non-neural cells that reside within or infiltrate into the injured area (including mast cells, basophils, platelets, macrophages, neutrophils, endothelial cells, keratinocytes, and fibroblasts). Collectively. these factors, referred to as the ‘inflammatory soup’, represent a wide array of signaling molecules, including neurotransmitters, peptides (substance P, CGRP, bradykinin), eicosinoids and related lipids (prostaglandins, thromboxanes, leukotrienes, endocannabinoids), neurotrophins, cytokines, and chemokines, as well as extracellular proteases and protons. Remarkably, nociceptors express one or more cell surface receptors capable of recognizing and responding to each of these pro-inflammatory or pro-algesic agents (Figure 4). Such interactions enhance excitability of the nerve fiber, thereby heightening its sensitivity to temperature or touch.
Unquestionably the most common approach to reducing inflammatory pain involves inhibiting the synthesis or accumulation of components of the inflammatory soup. This is best exemplified by non-steroidal anti-inflammatory drugs, such as aspirin or ibuprofen, which reduce inflammatory pain and hyperalgesia by inhibiting cyclooxygenases (Cox-1 and Cox-2) involved in prostaglandin synthesis. A second approach is to block the actions of inflammatory agents at the nociceptor. Here, we highlight examples that provide new insight into cellular mechanisms of peripheral sensitization, or which form the basis of new therapeutic strategies for treating inflammatory pain.
NGF is perhaps best known for its role as a neurotrophic factor required for survival and development of sensory neurons during embryogenesis, but in the adult, NGF is also produced in the setting of tissue injury and constitutes an important component of the inflammatory soup (Ritner et al., 2009). Among its many cellular targets, NGF acts directly on peptidergic C fiber nociceptors, which express the high affinity NGF receptor tyrosine kinase, TrkA, as well as the low affinity neurotrophin receptor, p75 (Chao, 2003; Snider and McMahon, 1998). NGF produces profound hypersensitivity to heat and mechanical stimuli through two temporally distinct mechanisms. At first, a NGF-TrkA interaction activates downstream signaling pathways, including phospholipase C (PLC), mitogen-activated protein kinase (MAPK), and phosphoinositide 3-kinase (PI3K). This results in functional potentiation of target proteins at the peripheral nociceptor terminal, most notably TRPV1, leading to a rapid change in cellular and behavioral heat sensitivity (Chuang et al., 2001).
Irrespective of their pro-nociceptive mechanisms, interfering with neurotrophin or cytokine signaling has become a major strategy for controlling inflammatory disease or resulting pain. The main approach involves blocking NGF or TNF-α action with a neutralizing antibody. In the case of TNF-α, this has been remarkably effective in the treatment of numerous autoimmune diseases, including rheumatoid arthritis, leading to dramatic reduction in both tissue destruction and accompanying hyperalgesia (Atzeni et al., 2005). Because the main actions of NGF on the adult nociceptor occur in the setting of inflammation, the advantage of this approach is that hyperalgesia will decrease without affecting normal pain perception. Indeed, anti-NGF antibodies are currently in clinical trials for treatment of inflammatory pain syndromes (Hefti et al., 2006).
Glutamate/NMDA Receptor-Mediated Sensitization
Acute pain is signaled by the release of glutamate from the central terminals of nociceptors, generating excitatory post-synaptic currents (EPSCs) in second order dorsal horn neurons. This occurs primarily through activation of postsynaptic AMPA and kainate subtypes of ionotropic glutamate receptors. Summation of sub-threshold EPSCs in the postsynaptic neuron will eventually result in action potential firing and transmission of the pain message to higher order neurons.
Other studies indicate that changes in the projection neuron, itself, contribute to the dis- inhibitory process. For example, peripheral nerve injury profoundly down-regulates the K+- Cl- co-transporter KCC2, which is essential for maintaining normal K+ and Cl- gradients across the plasma membrane (Coull et al., 2003). Downregulating KCC2, which is expressed in lamina I projection neurons, results in a shift in the Cl- gradient, such that activation of GABA-A receptors depolarize, rather than hyperpolarize the lamina I projection neurons. This would, in turn, enhance excitability and increase pain transmission. Indeed, pharmacological blockade or siRNA-mediated downregulation of KCC2 in the rat induces mechanical allodynia.

Sources:
Why does my shoulder hurt? A review of the neuroanatomical and biochemical basis of shoulder pain
Benjamin John Floyd Dean, Stephen Edward Gwilym, Andrew Jonathan Carr
Cellular and Molecular Mechanisms of Pain
Allan I. Basbaum1, Diana M. Bautista2, Grégory Scherrer1, and David Julius3
1Department of Anatomy, University of California, San Francisco 94158
2Department of Molecular and Cell Biology, University of California, Berkeley CA 94720 3Department of Physiology, University of California, San Francisco 94158
Molecular mechanisms of nociception
David Julius* & Allan I. Basbaum†
*Department of Cellular and Molecular Pharmacology, and †Departments of Anatomy and Physiology and W. M. Keck Foundation Center for Integrative Neuroscience, University of California San Francisco, San Francisco, California 94143, USA (e-mail: [email protected])
Post Disclaimer
Professional Scope of Practice *
The information on this blog site is not intended to replace a one-on-one relationship with a qualified healthcare professional or licensed physician and is not medical advice. We encourage you to make healthcare decisions based on your research and partnership with a qualified healthcare professional.
Blog Information & Scope Discussions
Welcome to El Paso's Premier Wellness and Injury Care Clinic & Wellness Blog, where Dr. Alex Jimenez, DC, FNP-C, a board-certified Family Practice Nurse Practitioner (FNP-BC) and Chiropractor (DC), presents insights on how our team is dedicated to holistic healing and personalized care. Our practice aligns with evidence-based treatment protocols inspired by integrative medicine principles, similar to those found on this site and our family practice-based chiromed.com site, focusing on restoring health naturally for patients of all ages.
Our areas of chiropractic practice include Wellness & Nutrition, Chronic Pain, Personal Injury, Auto Accident Care, Work Injuries, Back Injury, Low Back Pain, Neck Pain, Migraine Headaches, Sports Injuries, Severe Sciatica, Scoliosis, Complex Herniated Discs, Fibromyalgia, Chronic Pain, Complex Injuries, Stress Management, Functional Medicine Treatments, and in-scope care protocols.
Our information scope is limited to chiropractic, musculoskeletal, physical medicine, wellness, contributing etiological viscerosomatic disturbances within clinical presentations, associated somato-visceral reflex clinical dynamics, subluxation complexes, sensitive health issues, and functional medicine articles, topics, and discussions.
We provide and present clinical collaboration with specialists from various disciplines. Each specialist is governed by their professional scope of practice and their jurisdiction of licensure. We use functional health & wellness protocols to treat and support care for the injuries or disorders of the musculoskeletal system.
Our videos, posts, topics, subjects, and insights cover clinical matters and issues that relate to and directly or indirectly support our clinical scope of practice.*
Our office has made a reasonable effort to provide supportive citations and has identified relevant research studies that support our posts. We provide copies of supporting research studies available to regulatory boards and the public upon request.
We understand that we cover matters that require an additional explanation of how they may assist in a particular care plan or treatment protocol; therefore, to discuss the subject matter above further, please feel free to ask Dr. Alex Jimenez, DC, APRN, FNP-BC, or contact us at 915-850-0900.
We are here to help you and your family.
Blessings
Dr. Alex Jimenez DC, MSACP, APRN, FNP-BC*, CCST, IFMCP, CFMP, ATN
email: [email protected]
Licensed as a Doctor of Chiropractic (DC) in Texas & New Mexico*
Texas DC License # TX5807
New Mexico DC License # NM-DC2182
Licensed as a Registered Nurse (RN*) in Texas & Multistate
Texas RN License # 1191402
ANCC FNP-BC: Board Certified Nurse Practitioner*
Compact Status: Multi-State License: Authorized to Practice in 40 States*
Graduate with Honors: ICHS: MSN-FNP (Family Nurse Practitioner Program)
Degree Granted. Master's in Family Practice MSN Diploma (Cum Laude)
Dr. Alex Jimenez, DC, APRN, FNP-BC*, CFMP, IFMCP, ATN, CCST
(Board Certified: Family Practice Nurse Practitioner—Multistate)*
(Licensed Nurse Practitioner & Chiropractor - Multistate)*
Clinical Director
Digital Business Card
Dr. Maria Cardenas, MD
(Board Certified: Internal Medicine)
(Licensed Medical Doctor)
Medical Director, Clinical Director & Collaborative Physician
NPI # 1164426749
MD License #: J2933
Licenses and Board Certifications:
MD: Medical Doctor
DC: Doctor of Chiropractic
APRNP: Advanced Practice Registered Nurse
FNP-BC: Family Practice Specialization (Multi-State Board Certified)
RN: Registered Nurse (Multi-State Compact License)
CFMP: Certified Functional Medicine Provider
MSN-FNP: Master of Science in Family Practice Medicine
MSACP: Master of Science in Advanced Clinical Practice
IFMCP: Institute of Functional Medicine
CCST: Certified Chiropractic Spinal Trauma
ATN: Advanced Translational Neutrogenomics
Memberships & Associations:
TCA: Texas Chiropractic Association: Member ID: 104311
AANP: American Association of Nurse Practitioners: Member ID: 2198960
ANA: American Nurse Association: Member ID: 06458222 (District TX01)
TNA: Texas Nurse Association: Member ID: 06458222
NPI: 1205907805
| Primary Taxonomy | Selected Taxonomy | State | License Number |
|---|---|---|---|
| No | 111N00000X - Chiropractor | NM | DC2182 |
| Yes | 111N00000X - Chiropractor | TX | DC5807 |
| Yes | 363LF0000X - Nurse Practitioner - Family | TX | 1191402 |
| Yes | 363LF0000X - Nurse Practitioner - Family | FL | 11043890 |
| Yes | 363LF0000X - Nurse Practitioner - Family | CO | C-APN.0105610-C-NP |
| Yes | 363LF0000X - Nurse Practitioner - Family | NY | N25929 |
Dr. Alex Jimenez, DC, APRN, FNP-BC*, CFMP, IFMCP, ATN, CCST
(Board Certified: Family Practice Nurse Practitioner—Multistate)*
(Licensed Nurse Practitioner & Chiropractor - Multistate)*
Clinical Director
Digital Business Card
Dr. Maria Cardenas, MD
(Board Certified: Internal Medicine)*
(Licensed Medical Doctor)*
Medical Director, Clinical Director & Collaborative Physician
NPI # 1164426749
MD License #: J2933
Comments are closed.