

The Connection Between Inflammation and Depression

[et_pb_section bb_built=”1″][et_pb_row][et_pb_column type=”4_4″][et_pb_text]
One standard hypothesis of depression is that individuals who are depressed have a lack in monoamine receptors within the body, which in turn leads to reduced levels of neurotransmitters, such as serotonin and norephinephrine, in the brain. But growing evidence supports that at least some kinds of depression might also be linked to continuing low-grade inflammation in the body.
Previous research studies have linked depression with higher level of inflammatory markers when compared with people who aren’t depressed. When individuals are given pro-inflammatory cytokines, they report experiencing more symptoms of depression and anxiety. Chronically high levels of inflammation due to health issues, including chronic pain conditions, are also associated with high rates of depression. Even brain imaging of patients with depression show their brain scans have improved neuroinflammation. If your body is in an inflammatory state, fighting off the common cold or influenza, you can experience symptoms overlapping with depression, including, interrupted sleep, depressed mood, fatigue, foggy-headedness and impaired concentration.
A new study published in The Journal of Clinical Psychiatry supports the assumption that an increase in inflammation might play a role in depression. The huge study analyzed data from 14,275 people who have been interviewed between 2007 and 2012 using the Patient Health Questionnaire, or PHQ-9, to screen for depression and had blood samples drawn. They found that people who had depression had 46 percent greater levels of C-reactive protein, or CRP, a marker of inflammatory disease, in their own blood samples. The research study was just able to establish an association between inflammation and depression but not causation, even though it confirms the association of depression with high levels of inflammation as measured through CRP.
The theory that depression might be viewed as a psychoneuroimmunological disorder may also help explain why attempts to reduce chronic inflammation in the body also enhances and helps prevent depression. The purpose of the article below is to demonstrate as well as thoroughly discuss the role of inflammation in depression. Furthermore, the article will describe the evolutionary imperative to the modern treatment target, including a discussion of phytocannabinoids and their association with the treatment of a variety of health issues, including chronic pain symptoms.
Table of Contents
The Role of Inflammation in Depression: from Evolutionary Imperative to Modern Treatment Target
Abstract
Crosstalk between inflammatory pathways and neurocircuits in the brain can lead to behavioural responses, such as avoidance and alarm, that are likely to have provided early humans with an evolutionary advantage in their interactions with pathogens and predators. However, in modern times, such interactions between inflammation and the brain appear to drive the development of depression and may contribute to non-responsiveness to current antidepressant therapies. Recent data have elucidated the mechanisms by which the innate and adaptive immune systems interact with neurotransmitters and neurocircuits to influence the risk for depression. Here, we detail our current understanding of these pathways and discuss the therapeutic potential of targeting the immune system to treat depression.
Introduction
Depression is a devastating disorder, afflicting up to 10% of the adult population in the United States and representing one of the leading causes of disability worldwide1. Although effective treatments are available, approximately one third of all patients with depression fail to respond to conventional antidepressant therapies2, further contributing to the global burden of the disease. Accordingly, there is a pressing need for new conceptual frameworks for understanding the development of depression to develop better treatments. In this Review, we outline emerging data that point to the immune system — and, in particular, the inflammatory response — as a potentially important contributor to the pathophysiology of depression. We first consider the origins of this notion from an evolutionary perspective, examining the advantages of depressive behaviours in the context of host immune responses to pathogens, predators and conspecifics in ancestral environments. The pivotal role of psychosocial stress in the modern world are then examined, highlighting inflammasome activation and immune cell trafficking as novel mechanisms by which stress-induced inflammatory signals can be transmitted to the brain. Neurotransmitters and neurocircuits that are targets of the inflammatory response are also explored followed by an examination of brain–immune interactions as risk and resilience factors for depression. Finally, these interactions are discussed as a foundation for a new era of therapeutics that target the immune system to treat depression, with a focus on how immunological biomarkers can be used to personalize care.
An Evolutionary Perspective
Data from humans and laboratory animals provide compelling evidence that stress-relevant neurocircuitry and immunity form an integrated system that evolved to protect organisms from a wide range of environmental threats. For example, in the context of a laboratory stressor that entails delivering a speech to a judgmental panel of supposed ‘behavioural experts’, subjects experience a classic ‘fight or flight’ response characterized by increases in heart rate and blood pressure as well as in cortisol and catecholamines. But something else happens within the body that demands a deeper explanation. The stressor activates key inflammatory pathways in peripheral blood mononuclear cells, including activation of the transcription factor nuclear factor–κB (NF-κB), and leads to marked increases in circulating levels of pro-inflammatory cytokines, such as interleukin-6 (IL-6)3,4. In essence, the body mounts an immune response not against a pathogen, but against a threat to the subject’s self-esteem. Moreover, individuals at high risk of developing depression (for example, those who have experienced early-life trauma) show increased inflammatory responses to such laboratory stressors compared with low-risk individuals3. Furthermore, the greater the inflammatory response to a psychosocial stressor, the more probable the subject is to develop depression over the ensuing months5. Two questions immediately present themselves: why should a stimulus devoid of any pathogen induce an inflammatory response, and why should this response promote the development of depression?
Pathogen Host Defense and Depression
No coherent answer to these questions is apparent if immunity is viewed as merely another physiological system within the body. However, when seen against the back-drop of millions of years of co-evolution between mammals and the world of microorganisms and parasites, the human inflammatory bias exposed by laboratory stressors and reflected in the association between immune activation and depression not only makes imminent adaptive sense but also provides insight into a paradox deep within the heart of depression itself; namely, why are the genetic alleles that are most frequently associated with depression so common in the modern gene pool6 (FIG. 1)
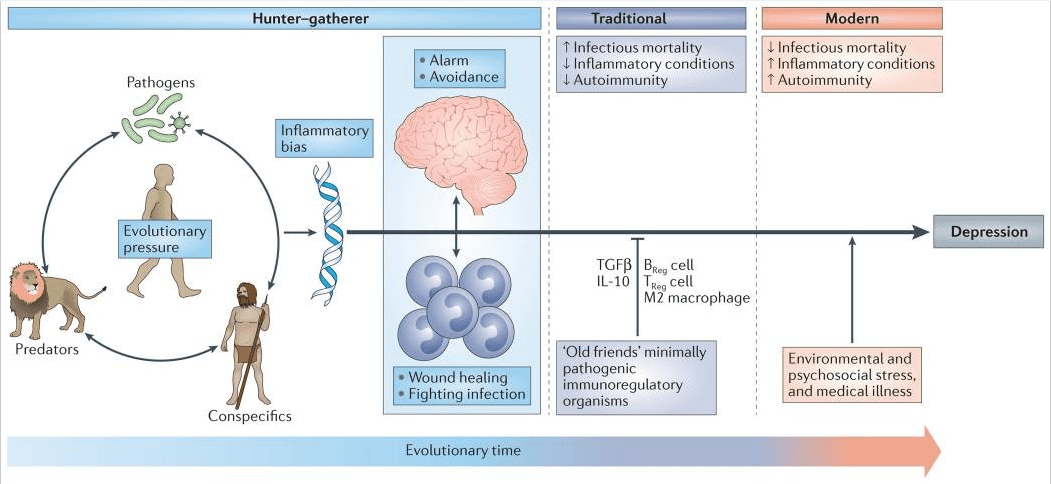
Most adaptive theories of depression have focused on the potential benefits of depressive symptoms for relationships with other humans7. However, recent models have shifted the focus away from relationships with people, to relationships — both detrimental and beneficial — with pathogens6,8. These theories, which are supported by converging evidence (BOX 1), posit that modern humans have inherited a genomic bias towards inflammation because this response — and the depressive symptoms it promotes — enhanced host survival and reproduction in the highly pathogenic environments in which humans evolved6. From this theoretical perspective, at least some of the human vulnerability to depression evolved out of a behavioural repertoire — often referred to as ‘sickness behaviour’ — which promoted host survival in the face of infection. Indeed, it has been hypothesized that the social avoidance and anhedonia characteristic of depression serve to shunt energy resources to fighting infection and wound healing, whereas the hypervigilance characteristic of anxiety disorders, commonly comorbid with depression, subserves protection from attack and subsequent pathogen exposure6,9. Even psychological stress can be understood from this theoretical perspective, given that the vast majority of stressors faced by mammals over evolutionary time boiled down to risks inherent in hunting, being hunted or competing for reproductive access or status. In all of these circumstances, the risk of pathogen invasion — and subsequent death from infection — was greatly increased as a result of wounding. In ancestral environments, the association between stress perception and risk of subsequent wounding was reliable enough that evolution favoured organisms that prepotently activated inflammatory systems in response to a wide array of environmental threats and challenges (including psychosocial stressors), even if this activation was often a ‘false alarm’ (REF. 6).
Pathogen Host Defense Hypothesis of Depression
Several lines of evidence support the notion that the evolution and persistence of depression risk alleles and depressive symptoms in human populations are based on their relevance to ‘pathogen host defence’. This evidence includes:
- Until recently, approximately 50% of humans died from infectious causes before adulthood, thereby providing strong selective pressure for genetic alleles that enhance host defence124.
- As a result of strong selective pressure, microbial interactions have been a primary driver of human evolution125.
- Patterns of inflammatory activation associated with depression promote survival in highly pathogenic environments while increasing mortality in sanitary conditions common in the developed world126.
- The best replicated risk alleles for depression have pro-inflammatory and/or anti-pathogen protective effects or have been implicated in social behaviours that are likely to reduce pathogen exposure6.
- Environmental risk factors for the development of depression (that is, psychosocial stress, early life adversity, obesity and processed-food diet) are uniformly pro-inflammatory13.
- Exposure to pro-inflammatory cytokines produces a sickness syndrome with symptoms that overlap considerably with those seen in depression and that can be ameliorated by treatment with antidepressants23. In addition, the onset of depression is often mistaken with development of sickness, and symptoms
- associated with infections are often mistaken with the onset of depression127.
- Chronic cytokine exposure produces a combination of withdrawal and/or energy conservation, anxiety and/or hypervigilance behaviours and emotions that commonly coexist in depression6,9.
- Symptoms shared by depression and sickness behaviour — such as hyperthermia and reduced iron availability — that lack any conceivable social value have potent anti-pathogen effects6.
The ‘pathogen host defence’ hypothesis of depression may also provide insight into the twofold increase in depression in women compared to men, especially during the reproductive years10. Recent data indicate that women are more sensitive to the behavioural effects of inflammation, demonstrating greater increases in depressed mood than men following endotoxin exposure despite a similar magnitude in cytokine (IL-6 and tumour necrosis factor (TNF)) responses11. Women also exhibit a greater likelihood than men to develop depression in response to standardized doses of interferon-α (IFNα)12. By being more sensitive to inflammation-induced depressive symptoms, women may have benefited more from the protection provided by these symptoms in terms of fighting infection, healing wounds and avoiding subsequent pathogen exposure. Given the potentially negative impact of inflammation on reproductive success (for example, by reducing fertility and impairing lactation), the increase of depressive symptoms in women across evolutionary time may have given women of reproductive age an advantage in coping with and avoiding pathogens and the related inflammation, with increased depressive disorders being the ultimate trade-off in modern times.
Modern Exaggeration of the Inflammatory Bias
The prevalence of autoimmune, allergic and inflammatory diseases has markedly increased in the past 100 years, and rates of these conditions follow a similar upward trajectory in societies transitioning from traditional (that is, rural) to modern (that is, urban) ways of life13. Increasing evidence suggests that this pattern of widespread immune dysregulation may result from disruptions in our relationship and/or contact with a variety of co-evolved, non-lethal immunoregulatory microorganisms and parasites, especially commensals and symbiotes in the microbiotas of the gut, skin and nasal and oral cavities, that were ubiquitous in the natural environments in which humans evolved14. Although widely disparate, these organisms (often referred to as ‘old friends’) share a tendency to reduce inflammation and suppress effector immune cells through the induction of IL-10 and transforming growth factor-β (TGFβ) while promoting the development of anti-inflammatory immune cell populations, such as alternatively activated (also referred to as ‘M2’) macrophages and regulatory T (TReg) cells and regulatory B cells13,14 (FIG. 1). Owing to various cultural changes, including the loss of exposure to microbial diversity with the advent of sanitation practices, modern humans now lack this immunoregulatory input — especially during infancy and childhood. Consequently, we find ourselves in a condition of an exacerbated inflammatory bias, with the particular conditions afflicting any given individual largely the result of genetic predisposition and environmental (for example, psychosocial) exposures13,14, ultimately accounting for the high co-morbidity between depression and autoimmune, allergic and inflammatory disorders13,15.
Inflammation and Depression
Data supporting the role of inflammation in depression are extensive and include findings that span experimental paradigms. Patients with major depressive disorder exhibit all of the cardinal features of an inflammatory response, including increased expression of pro-inflammatory cytokines and their receptors and increased levels of acute-phase reactants, chemokines and soluble adhesion molecules in peripheral blood and cerebrospinal fluid (CSF)16,17. Peripheral blood gene expression profiles consistent with a pro-inflammatory ‘M1’ macrophage phenotype and an over-representation of IL-6, IL-8 and type I IFN-induced signalling pathways have also been described18–20. In addition, increased expression of a variety of innate immune genes and proteins, including IL-1β, IL-6, TNF, Toll-like receptor 3 (TLR3) and TLR4, has been found in post-mortem brain samples from suicide victims that had depression16,18,19,21. Meta-analyses of the literature conclude that peripheral blood IL-1β, IL-6, TNF and C-reactive protein (CRP) are the most reliable biomarkers of inflammation in patients with depression16. Polymorphisms in inflammatory cytokine genes, including those encoding IL-1β, TNF and CRP, have also been associated with depression and its response to treatment22. Moreover, other genes implicated in depression derived from meta-analyses of genome-wide association studies have been linked to the immune response and the response to pathogens including TNF6 (BOX 1). Administration of inflammatory cytokines (for example, IFNα) or their inducers (for example, endotoxin or typhoid vaccination) to otherwise non-depressed individuals causes symptoms of depression23–26. Furthermore, blockade of cytokines, such as TNF, or of inflammatory signalling pathway components, such as cyclooxygenase 2, has been shown to reduce depressive symptoms in patients with medical illnesses, including rheumatoid arthritis, psoriasis and cancer, as well as in patients with major depressive disorder27–29.
As the field has matured, it has become increasingly apparent that inflammatory markers are elevated not only in a subgroup of patients with depression30,31 but also in patients with other neuropsychiatric disorders including anxiety disorders and schizophrenia32,33. Moreover, as described below, it may be more accurate to characterize the impact of inflammation on behaviour as being associated not wholly with depression but with specific symptom dimensions across diagnoses that align with the Research Domain Criteria framework put forth by the National Institute of Mental Health (US Department of Health and Human Services). These symptoms, including positive and negative valence systems, relate to altered motivation and motor activity (anhedonia, fatigue and psychomotor impairment) and increased threat sensitivity (anxiety, arousal and alarm)34. Finally, inflammation has been associated with antidepressant treatment non-responsiveness9,32,35–37. For example, in a recent study, 45% of patients with non-response to conventional antidepressants exhibited a CRP >3 mg L−1 (REF. 30), which is considered indicative of a high level of inflammation on the basis of widely accepted cut-off points38. Of note, however, the percentage of patients with high CRP levels can vary as a function of the population being studied, with higher percentages in patients with depression and treatment resistance, childhood maltreatment, medical illnesses and metabolic syndrome.
Immune Pathways Involved in Depression
Inflammasomes: Stress in Translation
Exposure to psychosocial stress is one of the most robust and reproducible predictors of developing depression in humans and is the primary experimental pathway to depressive-like behaviour in laboratory animals. Thus, the observation that exposure to a psychosocial laboratory stressor can activate an inflammatory response in humans was a major breakthrough in linking inflammation to depression3,4. An important question for the field, however, is by what mechanism is stress translated into inflammation? Although considerable attention has been paid to stress-induced neuroendocrine pathways, including the hypothalamic–pituitary–adrenal (HPA) axis and the sympathetic nervous system (SNS), both of which have immunomodulatory functions39, recent focus has been shifted towards inflammasomes, which may represent a crucial immunological interface between stress and inflammation40 (FIG. 2). Inflammasomes are cytosolic protein complexes that form in myeloid cells in response to pathogenic microorganisms and non-pathogenic or ‘sterile’ stressors. Assembly of the inflammasome leads to activation of caspase 1, which then cleaves the precursor forms of IL-1β and IL-18 into the active cytokines41. Given the relatively sterile nature of psychosocial stress, primary interest has been directed towards understanding how inflammasome activation in depression may be triggered by endogenous damage-associated molecular patterns (DAMPs), including ATP, heat shock proteins (HSPs), uric acid, high mobility group box 1 (HMGB1) and a variety of molecules linked with oxidative stress. Indeed, all of these DAMPs are induced by the psychological and mixed (that is, psychological and physiological) stressors used in animal models of depression42; an effect that is in part mediated by stress-induced release of catecholamines43. Moreover, studies in laboratory animals indicate that chronic mild-stress activates the NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3) inflammasome, which is well-known to respond to DAMPs44,45. Blockade of NLRP3 reverses stress-induced increases in IL-1β in the peripheral blood and brain, while also abrogating depressive-like behaviour in mice45. Interestingly, NLRP3 inflammasome upregulation and caspase-mediated cleavage of the glucocorticoid receptor can cause resistance to the effects of glucocorticoids, which are among the most potent anti-inflammatory hormones in the body46,47. Stress-induced glucocorticoid resistance is a well-characterized biological abnormality in patients with major depressive disorder and has been associated with increased inflammation48,49.
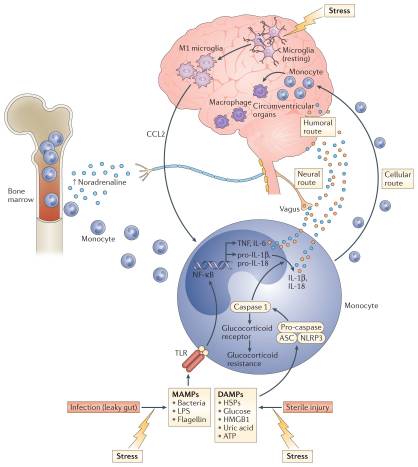
Supporting the potential role of the NLRP3 inflammasome in human depression are data demonstrating that increased expression of NLRP3 and caspase 1 in peripheral blood mononuclear cells of patients with depression is associated with increased blood concentrations of IL-1β and IL-18, which in turn correlate with depression severity19,50. In addition, DAMPs that are known to activate NLRP3 are increased in patients with mood disorders, with examples including HSPs, reactive oxygen species and other markers of oxidative stress such as xanthine oxidase, peroxides and F2-isoprostanes51–53. Finally, there is increasing interest in the potential role of the gut microbiome in mood regulation, which may be mediated in part by the inflammasomes54. Indeed, non-pathogenic commensal bacteria and derived microbial-associated molecular patterns (MAMPs) in the gut can leak into the peripheral circulation during stress and activate the inflammasomes55, a process mediated by the SNS and catecholamines56 (FIG. 2). Of note, stress-induced increases in IL-1β and IL-18 were attenuated by treating animals with antibiotics or neutralizing lipopolysaccharide (LPS), demonstrating the importance of the composition of the gut microbiome and gut permeability in stress-induced inflammatory responses55. Taken together, these data support the notion that the inflammasome may be a key immunological point of integration of stress-induced danger signals that ultimately drive inflammatory responses relevant to depression.
Transmitting Inflammatory Signals to the Brain
In addition to increased expression of innate immune cytokines and TLRs in post-mortem brain samples from suicide victims with depression, evidence of microglial and astroglial activation in several brain regions including frontal cortex, anterior cingulate cortex (ACC) and thalamus in post-mortem studies of patients with depression have been described57–59,60. Moreover, a well-controlled neuroimaging study using positron emission tomography (PET) and a radiolabelled tracer for the translocator protein (TSPO) — which is overexpressed in activated microglia, macrophages and astrocytes — revealed increased immune activation in the brains of patients with major depressive disorder compared with control subjects61. Of note, not all studies have revealed increased TSPO binding in patients with depression, possibly owing to effects of medication and/or a paucity of subjects with increased inflammation61,62. However, data from endotoxin administration to healthy volunteers indicates that radiolabelled TSPO ligands can readily identify cellular activation in several regions of the brain following a potent peripheral immune stimulus63.
Work from laboratory animal studies has elucidated several pathways through which inflammatory signals can be transmitted from the periphery to the brain (FIG. 2). These data support the idea that inflammatory responses in peripheral tissues may drive inflammation in the brain leading to depression. Much of the early work focused on how inflammatory cytokines, which are relatively large molecules, could cross the blood–brain barrier (BBB) and influence brain function64. Two major pathways have been described: the ‘humoral pathway’, which involves cytokine passage through leaky regions in the BBB, such as the circumventricular organs, and the binding of cytokines to saturable transport molecules on the BBB; and the ‘neural pathway’, which involves the binding of cytokines to peripheral afferent nerve fibres, such as the vagus nerve, that in turn stimulate ascending catecholaminergic fibres in the brain and/or are translated back into central cytokine signals16. More recently, however, attention has shifted to a third pathway referred to as the ‘cellular pathway’, which involves the trafficking of activated immune cells, typically monocytes, to the brain vasculature and parenchyma. The details of this pathway have been elegantly dissected in the context of behavioural changes in mice that are associated with peripherally induced inflammation in the liver65. In these studies, the release of TNF from inflamed liver was found to stimulate microglial cell production of CC-chemokine ligand 2 (CCL2; also known as MCP1) that then attracted monocytes to the brain65. Blockade of monocyte infiltration to the brain using antibodies specific for the adhesion molecules P-selectin and α4 integrin abrogated depressive-like behaviour in this animal model65. Of note, cytokine-stimulated astrocytes also may be major producers of chemokines, such as CCL2 and CXC-chemokine ligand 1 (CXCL1), that attract immune cells to the brain66. The cellular pathway additionally has been elucidated in the context of social defeat stress, whereby GFP-labelled monocytes coalesced in several regions of the brain associated with the detection of threat (for example, amygdala) — an effect that was dependent on CCL2 and was facilitated by mobilization of monocytes from the bone marrow as a result of stress-induced release of catecholamines67,68 (FIG. 2). Of note, initial microglial activation during social defeat stress appeared to be a result of neuronal activation by catecholamines and decreased neuronal production of CX3C-chemokine ligand 1 (CX3CL1; also known as fractalkine), which maintains microglia in a quiescent state67,68. Interestingly, this cellular pathway has received intriguing support from post-mortem analyses of brain tissue from patients with depression who committed suicide that showed increased numbers of perivascular macrophages in association with increased gene expression of allograft inflammatory factor 1 (AIF1, also known as IBA1) and CCL2, which are associated with macrophage activation and cellular trafficking59.
This evidence of peripheral myeloid cells trafficking to the brain during depression constitutes some of the first data supporting the existence of a central inflammatory response in human depression that is primarily driven by peripheral inflammatory events. Moreover, data demonstrate that antibodies that are specific for TNF but which do not cross the BBB, can block stress-induced depression in mice69. These findings indicate that peripheral inflammatory responses not only can provide important clues to the immunological mechanisms of inflammation in depression but also may serve as biomarkers and targets of immune-based therapies for depression. Protein biomarkers such as plasma CRP and TNF as well as immunotherapies targeting individual cytokines such as TNF, IL-1 and IL-6 may be most relevant in this regard. Of note, plasma CRP is a strong response predictor in anti-cytokine therapy70.
Cytokines and Neurotransmitters
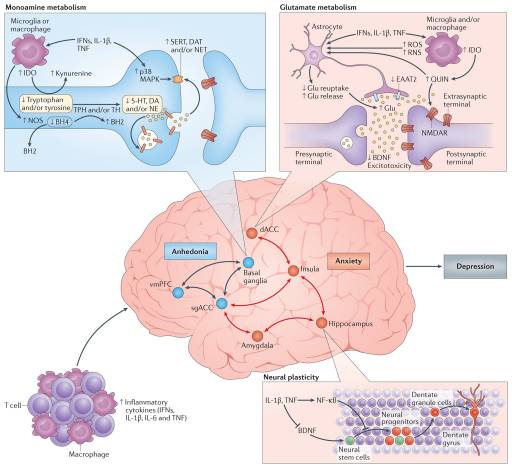
Given the pivotal importance of neurotransmission to mood regulation, attention has been paid to the impact of inflammation and inflammatory cytokines on the monoamines serotonin, noradrenaline and dopamine, as well as on the excitatory amino acid glutamate (FIG. 3). There are several pathways through which inflammatory cytokines can lead to reduced synaptic availability of the monoamines, which is believed to be a fundamental mechanism in the pathophysiology of depression71. For example, IL-1β and TNF induction of p38 mitogen-activated protein kinase (MAPK) has been shown to increase the expression and function of the reuptake pumps for serotonin, leading to decreased synaptic availability of serotonin and depressive-like behaviour in laboratory animals72. Through the generation of reactive oxygen and nitrogen species, inflammatory cytokines have also been found to decrease the availability of tetrahydrobiopterin (BH4), a key enzyme co-factor in the synthesis of all monoamines that is highly sensitive to oxidative stress73. Indeed, CSF concentrations of BH4 have been shown to be negatively correlated with CSF levels of IL-6 in patients treated with the inflammatory cytokine IFNα74. In addition, the plasma phenylalanine to tyrosine ratio, an indirect measure of BH4 activity, was shown to correlate with CSF concentrations of dopamine as well as symptoms of depression in IFNα-treated patients74. Activation of the enzyme indoleamine 2,3-dioxygenase (IDO) is also believed to be involved in cytokine-induced neurotransmitter alterations, in part by diverting the metabolism of tryptophan (the primary amino acid precursor of serotonin) into kynurenine, a compound that can be converted into the neurotoxic metabolite quinolinic acid by activated microglia and infiltrating monocytes and macrophages in the brain75,76. Of note, increased levels of quinolinic acid have been found in microglia in the ACC of suicide victims who suffered from depression77. Quinolinic acid directly activates receptors for glutamate (that is, N-methyl-D-aspartate (NMDA) receptors) while also stimulating glutamate release and blocking glutamate reuptake by astrocytes78. The effects of quinolinic acid on glutamate converge with the direct effects of pro-inflammatory cytokines on glutamate metabolism that include decreasing the expression of astrocyte glutamate reuptake pumps and stimulating astrocytic glutamate release79, ultimately contributing to excessive glutamate both within and outside the synapse. The binding of glutamate to extrasynaptic NMDA receptors leads to increased excitotoxicity and decreased production of brain-derived neurotrophic factor (BDNF)80. BDNF fosters neurogenesis, an important prerequisite for an antidepressant response, and has been shown to be reduced by IL-1β and TNF and their downstream signalling pathways including NF-κB in stress-induced animal models of depression81,82. Increased levels of glutamate in the basal ganglia and dorsal ACC (dACC) — as measured by magnetic resonance spectroscopy (MRS) — have been described in patients receiving IFNα, and higher levels of glutamate correlated with an increase in depressive symptoms83. More recent data indicate that in patients with depression, increased inflammation as reflected by a CRP >3 mg L−1 is also associated with increased basal ganglia glutamate (compared with patients with a CRP <1 mg L−1) that correlated with anhedonia and decreased psychomotor speed84. Interestingly, blocking glutamate receptors with ketamine or inhibiting IDO activity protects mice from LPS- or stress-induced depressive-like behaviour but leaves the inflammatory response intact85,86. These results indicate that increased activation of glutamate receptors by glutamate and/or quinolinic acid may be a common pathway through which inflammation causes depressive-like behaviour, suggesting that drugs that block glutamate receptor signalling and/or activation of the IDO pathway and its downstream metabolites might have unique applicability to patients with depression and increased inflammation. Importantly, conventional antidepressant medications act by increasing synaptic availability of monoamines and increasing neurogenesis through induction of BDNF87. Therefore, cytokines such as IL-1β and TNF serve to undermine these activities as they decrease the synaptic availability of monoamines while also decreasing BDNF and increasing extracellular glutamate, which is not a target of conventional antidepressant therapy. These cytokine-driven effects may explain the observations that increased inflammation is associated with less robust antidepressant treatment responses and that treatment-resistant patients exhibit increased inflammatory markers88.
Effects of Inflammation on Neurocircuitry
Given the impact of cytokines on neurotransmitter systems that regulate the functional activity of neurocircuits throughout the brain, it is no surprise that neuroimaging studies have revealed cytokine-induced alterations in regional brain activity. Consistent with the evolutionary advantages of the partnership between the brain and the immune system, primary cytokine targets in the CNS involve those brain regions that regulate motivation and motor activity (promoting social avoidance and energy conservation) as well as arousal, anxiety and alarm (promoting hypervigilance and protection against attack) (FIG. 3).
Dopamine has a fundamental role in motivation and motor activity, and cytokines have been shown to decrease the release of dopamine in the basal ganglia in association with decreased effort-based motivation as well as reduced activation of reward circuitry in the basal ganglia, in particular the ventral striatum89–91. Inflammatory stimuli have been associated with reductions in reward responsiveness in the striatum across many neuroimaging platforms, demonstrating the validity and reproducibility of these cytokine-mediated effects on the brain in otherwise non-depressed individuals peripherally administered IFNα, endotoxin or typhoid vaccination and imaged by PET, functional magnetic resonance imaging (fMRI), MRS and quantitative magnetization transfer imaging83,89,90,92,93. Interestingly, recent fMRI studies suggest that inflammation-induced decreases in responsiveness to positive reward are also associated with increased sensitivity to aversive stimuli (that is, negative reinforcement) and reduced responsiveness to novelty in the substantia nigra (which is another dopamine-rich structure in the basal ganglia)93,94. Typhoid vaccination has also been shown to activate the subgenual ACC (sgACC), a brain region implicated in depression, and to decrease connectivity of the sgACC with the ventral striatum, an effect modulated by plasma IL-6 (REF. 26). These fMRI findings have recently been extended to patients with depression whose increased plasma CRP level is associated with decreased functional connectivity within reward-related circuits including the ventral striatum and the ventromedial prefrontal cortex that, in turn, mediates the relationship between CRP and anhedonia95. Indeed, patients with depression with a CRP >3 mg L−1 had little, if any, connectivity within reward-related circuits as measured by fMRI, whereas connectivity in patients with depression with a CRP <1 mg L−1 was similar to healthy controls95. Taken together, these data support the notion that the effect of cytokines on the brain in general and dopaminergic pathways in particular lead to a state of decreased motivation or anhedonia, which is a core symptom of depression.
fMRI studies have demonstrated that increased inflammation is also associated with increased activation of threat- and anxiety-related neurocircuitry, including the dACC as well as the insula and amygdala26,96,97. Of note, the dACC and amygdala are regions that exhibit increased activity in patients with high-trait anxiety and neuroticism98, conditions that often accompany depression and are associated with increased inflammation. For example, increased concentrations of oral IL-6 and soluble TNF receptor 2 (also known as TNFRSF1B) in response to a public speaking stressor was significantly correlated with the response of the dACC to a social rejection task97. In addition, increased oral IL-6 expression in response to a social evaluation stressor was significantly correlated with activation of the amygdala, with subjects who exhibited the highest IL-6 responses to stress demonstrating the greatest connectivity within threat circuitry, including the amygdala and the dorsomedial prefrontal cortex, as measured by fMRI99. Interestingly, these data are consistent with the trafficking of monocytes to the amygdala during social defeat stress in mice68.
Risk and Resilience
Increased Inflammation and the Risk for Depression
Consistent with the emerging recognition that inflammation may cause depression in certain subgroups of individuals, epidemiological studies on large community samples — as well as smaller samples of medically ill individuals — have demonstrated that increased inflammation serves as a risk factor for the future development of depression. For example, increased peripheral blood CRP and IL-6 concentrations were found to significantly predict depressive symptoms after 12 years of follow up in the Whitehall II study of over 3,000 individuals, whereas no association was found between the presence of depressive symptoms and subsequent blood CRP and IL-6 levels100. Similar findings were reported in the English Longitudinal Study of Ageing in which a CRP >3 mg L−1 predicted depressive symptoms and not vice versa101. Of note, however, some studies have found no longitudinal relationship between depression and inflammation, and others have found that depression leads to increased inflammation102. Other factors known to be associated with increased peripheral inflammation, including childhood and adult trauma, have also been shown to be predictive of a greater risk of developing depression103,104.
Both genetic and epigenetic mechanisms may explain why childhood or adult traumas can contribute to exaggerated or persistent inflammation and, ultimately, depression. For example, polymorphisms in CRP were associated not only with increased peripheral blood concentrations of CRP but also with symptoms of post-traumatic stress disorder, especially heightened arousal, in individuals exposed to civilian trauma32. Moreover, gene–environment interactions have been found to influence depression severity in response to chronic interpersonal stress: individuals carrying polymorphisms in IL1B that are associated with higher expression of peripheral IL-1β exhibited more severe depressive symptoms in the context of interpersonal stress than individuals without the IL1B risk allele105. Similarly, mice in which peripheral blood leukocytes produced high concentrations of LPS-induced IL-6 ex vivo before stress exposure showed decreased social exploration after social defeat stress, whereas mice that produced low levels of IL-6 before stress exposure exhibited no behavioural effects in response to social defeat88. Of note, adoptive transfer of bone marrow progenitor cells from mice producing high levels of IL-6 ex vivo to mice that produced low levels of IL-6 made these formerly stress-resilient animals sensitive to the depressive effects of social defeat88.
Epigenetic changes in genes related to inflammation may also affect the risk for depression and anxiety in the context of psychosocial stress. Indeed, the well-documented association of childhood trauma with increased inflammation is linked to stress-induced epigenetic changes in FKBP5, a gene implicated in the development of depression and anxiety as well as in the sensitivity to glucocorticoids106. Allele-specific, childhood trauma-dependent DNA demethylation in functional glucocorticoid response elements of FKBP5 were found to be associated with decreased sensitivity of peripheral blood immune cells to the inhibitory effects of the synthetic glucocorticoid dexamethasone on LPS-induced production of IL-6 in vitro106. Of note, decreased activation of glucocorticoid receptor-responsive genes in association with increased activation of genes regulated by NF-κB has been found to be a ‘fingerprint’ of the effects of chronic stress in several studies examining a variety of psychosocial stressors39,107.
T Cells and Resilience to Depression
Some of the most intriguing data regarding the role of the immune system in depression come from studies showing that T cells may protect against stress and depression in laboratory animals. For example, the adoptive transfer of T cells from animals exposed to chronic social defeat stress led to an antidepressant behavioural phenotype in stress-naive mice, which was associated with decreased pro-inflammatory cytokines in serum, a shift towards a neuroprotective M2 phenotype in microglia and increased neurogenesis in the hippocampus108. Similar results have been reported following acute stress in mice, in which effector T cell migration to the choroid plexus as a result of glucocorticoid induction of intercellular adhesion molecule 1 (ICAM1) expression in the choroid plexus was associated with reduced anxiety-like behaviour109. Mice with impaired release of glucocorticoids in response to stress were anxiety prone109. Immunization of anxiety-prone animals with a CNS-specific antigen restored T cell trafficking to the brain during stress and reversed anxiety-like behaviour in association with increased neurogenesis109. Immunization with a CNS-specific antigen also blocked stress-induced depression in mice110. The mechanism by which T cells influence resilience is believed to be related to their production of IL-4 within the meningeal space. Through as yet uncharacterized pathways, IL-4 then stimulates astrocytes to produce BDNF, and also promotes the conversion of meningeal monocytes and macrophages from a pro-inflammatory M1 phenotype to a less inflammatory M2 phenotype111. The movement of T cells throughout the brain, including the meningeal space, has become an area of special interest with the recent description of a brain lymphatic system that heretofore had gone unrecognized112. Data also indicate that TReg cells may have a role in reducing inflammation and supporting neuronal integrity during stress113. Similar reports have characterized T cells that are activated by vagal nerve stimulation to produce acetylcholine, which can inhibit NF-κB activation by binding to the α7 subunit of the nicotinic acetylcholine receptor114.
Relevant to depression, however, peripheral T cell trafficking in response to glucocorticoids has been shown to be impaired in patients with depression, possibly owing to glucocorticoid resistance as a result of genetically mediated (for example, FKBP5) or inflammasome-mediated mechanisms targeting the glucocorticoid receptor46,115. In addition, inflammatory cytokines and their signalling pathways, including p38 MAPK, have direct inhibitory effects on glucocorticoid receptor function116. Moreover, patients with depression have been shown to have increased numbers of peripheral blood myeloid-derived suppressor cells, which inhibit T cell function117. Of note, activation of the NLRP3 inflammasome leads to increased accumulation of myeloid-derived suppressor cells118. Decreased numbers of peripheral blood TReg cells and reduced concentrations of anti-inflammatory cytokines in the blood, including TGFβ and IL-10, have also been reported in depression119. Thus, it appears that patients with depression may have impairments in neuroprotective and anti-inflammatory T cell responses.
These findings suggest that therapies that boost such T cell responses could be used in patients with depression. Examples include immunization strategies (with CNS antigens as discussed above) that attract T cells to the brain or administration of bacteria, such as Mycobacterium vaccae, or parasites that stimulate TReg cell responses or T cell production of IL-4 (REFS 14,109,110,120). Indeed, colonization of pregnant dams with helminths attenuated the increase of hippocampal IL-1β in neonatal rats infected with bacteria and protected these animals from the subsequent development of microglial sensitization and cognitive dysfunction in adulthood. This effect was associated with increased ex vivo production of IL-4 and decreased production of IL-1β and TNF by splenic macrophages in response to LPS stimulation120. Finally, vagus nerve stimulation could be used to induce anti-inflammatory acetylcholine-producing T cells121. Although many strategies exist to activate anti-inflammatory T cell responses including the induction of TReg cells by administration of mesenchymal stem cells122, the majority of the approaches discussed above have proof-of-concept data in animal models of depression. Nevertheless, the clinical relevance of these approaches has yet to be determined by randomized clinical trials in patients with depression.
Translational Considerations
Our increasing understanding of how inflammatory processes contribute to depression, combined with the growing frustration over the lack of discovery of new antidepressants, have stimulated interest in the possibility that various classes of anti-inflammatory medications or other anti-inflammatory strategies (as discussed above) may hold promise as novel ‘all-purpose’ antidepressants. Unfortunately, it appears that anti-inflammatory agents may only demonstrate effective antidepressant activity in subgroups of patients who show evidence of increased peripheral inflammation, for example individuals with medical conditions including osteoarthritis and psoriasis that are characterized by increased levels of peripheral inflammation and patients with depression with increased inflammatory markers29,30. Moreover, in patients with depression who do not show elevated peripheral levels of inflammation, anti-inflammatory treatments may actually impair placebo responses that contribute to the effectiveness of all known antidepressant modalities123. In the only study to date examining the antidepressant effect of a cytokine antagonist in medically healthy adults with treatment-resistant depression, post hoc analysis revealed a dose-response relationship between baseline levels of peripheral inflammation and subsequent antidepressant response to the TNF inhibitor infliximab30. In patients with baseline plasma CRP concentrations ≥5 mg L−1, infliximab outperformed placebo with an effect size similar to that observed in studies of standard antidepressants. Patients with a CRP >3 mg L−1, the standard cut-off for high inflammation, also exhibited separation from placebo. Of note, this latter finding along with data demonstrating the relevance of a CRP >3 mg L−1 to altered reward circuitry and glutamate metabolism in depression as well as the prediction of subsequent depressive episodes (described above) aligns well with other diseases in which a CRP >3 mg L−1 is relevant to prediction and pathology including cardiovascular disease and diabetes. These data suggest that the cut-off for high inflammation in depression may be consistent with other disorders (BOX 2). Importantly, however, in patients with lower levels of inflammation, blockade of TNF with infliximab actually impaired the placebo response30, suggesting that anti-inflammatory treatments in patients without inflammation may be detrimental, highlighting the growing recognition that the immune system has an important role in several processes central to neuronal integrity.
Guidelines for Anti-Inflammatory Clinical Trials in Depression
Based on the animal and human literature on the effects of cytokines on the brain, the following guidelines can inform clinical trials designed to test the cytokine hypothesis of depression.
- Inflammation only occurs in subgroups of patients with depression30. Clinical trials should enrich for patient populations with evidence of increased inflammation, particularly those identified by a C-reactive protein (CRP) >3 mg L−1, which has been shown to characterize patients with depression with altered reward circuitry and increased basal ganglia glutamate, as well as those who have shown a response to anti-cytokine therapy30,84,95.
- Anti-inflammatory drugs may harm patients without increased inflammation. Inflammatory cytokines and the innate immune response have pivotal roles in synaptic plasticity, neurogenesis, long-term potentiation (which is a fundamental process in learning and memory) and possibly antidepressant response123,128.
- Primary behavioural outcome variables should include measures of anhedonia and anxiety. Neuroimaging studies coupled with studies administering a variety of inflammatory stimuli, including the inflammatory cytokine interferon-α, endotoxin and typhoid vaccination, have revealed that inflammation targets neurocircuits in the brain that regulate motivation and reward as well as anxiety, arousal and alarm35. In addition, these symptoms have been shown to respond to anti-cytokine therapy in limited studies.
- Drugs that specifically target inflammatory cytokines and/or their signalling pathways are preferable. The majority of clinical trials to date have used anti-inflammatory drugs (non-steroidal anti-inflammatory agents and minocycline, a tetracycline antibiotic) that have several off-target effects making the extant data relevant to testing the cytokine hypothesis of depression difficult to interpret31.
- Target engagement must be established in the periphery and ultimately the brain. Protein and gene expression markers of inflammation in the peripheral blood can serve as relevant proxies for inflammation in the brain129, especially given evidence of trafficking of activated peripheral immune cells to the brain in stress-induced animal models of depression. Relevant therapeutic interventions should decrease peripheral inflammatory markers in concert with improvement of specific depressive symptoms. Translocator protein neuroimaging ligands may ultimately serve as direct measures of neuroinflammation and its inhibition by anti-inflammatory therapies in future clinical trials61.
We conclude by offering the balanced perspective that anti-inflammatory therapies are unlikely to be all-purpose antidepressants. Perhaps we only think of standard antidepressants as all-purpose agents because we have never succeeded in developing predictive biomarkers that would reliably inform us of who will respond to any given agent. If so, then we view these agents as all-purpose, not because it is true but out of hope and ignorance. Thus, instead of being a negative, perhaps the finding that baseline inflammatory biomarkers such as CRP can predict subsequent symptomatic response to anti-inflammatory strategies is, in fact, the most positive development thus far in our quest to understand how the immune system might be harnessed to improve the treatment of depression.

Dr. Alex Jimenez’s Insight
When you catch a cold, certain symptoms are triggered by inflammatory markers released in response to illness. While sneezing, coughing and a sore throat serve as the most “obvious” signs you may be sick, what truly keeps you in bed when you have a cold is the accompanying fatigue, inattentiveness, loss of appetite, change in sleep pattern, heightened perception of pain and apathetic withdrawal. These symptoms are similar to the wide array of symptoms that define depression. Many research studies have demonstrated that depression may occur due to an inflammatory response to illness, just as in the case of a common cold. The connection between inflammation and depression has long been argued among healthcare professionals and researchers, where new evidence could open the doors to additional treatment approaches which could help better manage this debilitation health issue.
Conclusion
In ancestral times, integration of inflammatory responses and behaviours of avoidance and alarm provided an evolutionary advantage in managing the microbial world. In the absence of the temporizing influence of commensal organisms that were rife in environments in which humans evolved, the inflammatory bias of the human species in the civilized world has been increasingly engaged in the complex world of psychosocial interactions and the inevitable stress it engenders. Responding to these sterile insults with activation of the inflammasome and mobilization of myeloid cells to the brain, the resultant release of inflammatory cytokines impinges on neurotransmitters and neurocircuits to lead to behaviours that are poorly suited for functioning in modern society. This inevitability of our evolutionary past is apparent in the high rates of depression that are seen in society today. There is also an increasing recognition of mechanisms of resilience that derive from our emerging understanding of the neuroprotective effects of a variety of T cell responses ranging from effector T cells that produce IL-4 to TReg cells with anti-inflammatory properties. A better understanding of these neuroprotective pathways and of the inflammatory mechanisms — from inflammasome activation to cell trafficking to the brain — that operate in patients with depression may lead to the development of novel anti-depressant therapies.
For glossary and footnotes visit: Ncbi.nlm.nih.gov/pmc/articles/PMC5542678/
Understanding the Phytocannabinoids
The discovery of the body’s endocannabinoid system, or ECS, in the 1980s provided researchers a totally new outlook on the chemicals in marijuana and hemp that had formerly been identified 40 years earlier, including how these chemicals interacted with and acted on a prevalent regulatory system in the human body. The title given to those chemicals was phytocannabinoids, meaning “phyto” for plantlife. Over 80 phytocannabinoids are identified in marijuana and hemp. The psychoactive phytocannabinoid in marijuana, tetrahydrocannabinol, or THC, represents only one of many phytocannabinoids now being extensively studied for its numerous health benefits. The more science learns about the far-reaching effects of the ECS in encouraging brain health, in improving immune function, in keeping a healthy inflammatory response, and in promoting GI health, fertility, bone health, and much more, the more interest there is in locating these phytocannabinoids in nature and learning how they affect human health. Due to this widespread interest, phytocannabinoids have been identified in many plants outside the Cannabis species; for instance, plants such as clove, black pepper, Echinacea, ginseng, broccoli, and carrots, all contain phytocannabinoids.
Phytocannabinoids in Hemp
Although the majority of people have now heard of cannabadiol (CBD), it’s only one of lots of the constituents in hemp which interact with the ECS. Two other notable phytocannabinoids include:
Cannabichromene (CBC)
CBC was first analyzed in the 1980s as it was found to modulate a normal inflammatory response in a rat model. More recently CBC has been shown to promote brain health, skin health, and preserve normal motility in the digestive tract.
Cannabigerol (CBG)
CBG has been increasingly studied for its capacity to support nervous system health. CBG has multiple roles from the ECS, such as inhibiting the reuptake of anandamide, an extremely beneficial endocannabinoid we make within our own bodies. CBG might also provide help for immune function, skin health, and a positive disposition. CBG is typically found in much higher concentrations in industrial hemp than in marijuana.
Phytocannabinoids in Other Crops
There is also ongoing research in discovering phytocannabionids in many other plants. Some of them include:
Beta-Caryophyllene (BCP)
Although BCP is located from the flowers and leaves of hemp, since just the hemp stalk is used in nutritional supplements, even BCP content is lost. But, BCP is contained in many other plants, like cloves and black pepper. BCP binds to the CB2 cannabinoid receptor in the body, and by doing so it helps maintain a healthy inflammatory reaction and promotes the overall health of the digestive tract, skin, and liver disease.
Diindolylmethane (DIM)
DIM is a compound we create in our bodies when we eat cruciferous vegetables like broccoli, cauliflower, cabbage, and Brussels sprouts. DIM is also a readily available nutritional supplement. Much like beta-caryophyllene, DIM binds to the CB2 cannabinoid receptor. Since the immune system is rich with CB2 receptors, this may clarify the immune-supportive health benefits of the foods.
Alkylamides
Located in the familiar herb Echinacea, alkylamides are also drawing interest for their part in the ECS. These unique compounds act on the CB2 cannabinoid receptor to regulate cytokine synthesis and also to support immune function. This activity probably helps clarify some of the common uses of Echinacea.
Falcarinol
Found in carrots, celery, parsley, and Panax ginseng, this interesting compound may not be one need to touch. Falcarinol binds to the CB1 cannabinoid receptor which also has the reverse effect of anandamide, that the cannabinoid our bodies make that binds to the receptor. Because of this trend, falcarinol can cause an allergic skin reaction that’s regarded because it prevents our very own ECS from modulating local inflammation.
Yangonin
This phytocannabinoid, found from the Kava plant (Piper methysticum), binds to CB1 cannabinoid receptors and also acts on GABA receptors in the nervous system. Although yangonin appears to promote relaxation and modulate responses to stress, it might also be bad for the liver.
Understanding of the endocannabinoid system is growing quickly. As this knowledge does enlarge, science will continue to find more phytocannabinoids in plants and foods that are useful in supporting health in many ways.
In conclusion, many research studies have found a connection between inflammatory pathways and neurocircuits in the brain which can lead to a variety of behavioral responses, such as avoidance and alarm, however, growing evidence has demonstrated that chronic inflammation can lead to depression. Depression is a debilitating disorder which amounts to one of the leading causes of disability worldwide. The article above describes the connection between inflammation and depression. New insights on the results of the research studies could open the possibilities for new treatments to treat depression, among other associated health issues. Moreover, understanding the role of phytocannabinoids in the human body can function as another treatment approach for inflammation associated with depression. Information referenced from the National Center for Biotechnology Information (NCBI). The scope of our information is limited to chiropractic as well as to spinal injuries and conditions. To discuss the subject matter, please feel free to ask Dr. Jimenez or contact us at 915-850-0900 .
Curated by Dr. Alex Jimenez

Additional Topics: Back Pain
Back pain is one of the most prevalent causes for disability and missed days at work worldwide. As a matter of fact, back pain has been attributed as the second most common reason for doctor office visits, outnumbered only by upper-respiratory infections. Approximately 80 percent of the population will experience some type of back pain at least once throughout their life. The spine is a complex structure made up of bones, joints, ligaments and muscles, among other soft tissues. Because of this, injuries and/or aggravated conditions, such as herniated discs, can eventually lead to symptoms of back pain. Sports injuries or automobile accident injuries are often the most frequent cause of back pain, however, sometimes the simplest of movements can have painful results. Fortunately, alternative treatment options, such as chiropractic care, can help ease back pain through the use of spinal adjustments and manual manipulations, ultimately improving pain relief.

EXTRA IMPORTANT TOPIC: Lower Back Pain Management
MORE TOPICS: EXTRA EXTRA: Chronic Pain Care Center
[/et_pb_text][et_pb_accordion _builder_version=”3.9″][et_pb_accordion_item _builder_version=”3.9″ title=”Blank” use_background_color_gradient=”off” background_color_gradient_start=”#2b87da” background_color_gradient_end=”#29c4a9″ background_color_gradient_type=”linear” background_color_gradient_direction=”180deg” background_color_gradient_direction_radial=”center” background_color_gradient_start_position=”0%” background_color_gradient_end_position=”100%” background_color_gradient_overlays_image=”off” parallax=”off” parallax_method=”on” background_size=”cover” background_position=”center” background_repeat=”no-repeat” background_blend=”normal” allow_player_pause=”off” background_video_pause_outside_viewport=”on” text_shadow_style=”none” box_shadow_style=”none” custom_css_main_element=”display:none;” /][et_pb_accordion_item _builder_version=”3.9″ title=”References” use_background_color_gradient=”off” background_color_gradient_start=”#2b87da” background_color_gradient_end=”#29c4a9″ background_color_gradient_type=”linear” background_color_gradient_direction=”180deg” background_color_gradient_direction_radial=”center” background_color_gradient_start_position=”0%” background_color_gradient_end_position=”100%” background_color_gradient_overlays_image=”off” parallax=”off” parallax_method=”on” background_size=”cover” background_position=”center” background_repeat=”no-repeat” background_blend=”normal” allow_player_pause=”off” background_video_pause_outside_viewport=”on” text_shadow_style=”none” box_shadow_style=”none”]
[/et_pb_accordion_item][et_pb_accordion_item _builder_version=”3.9″ title=”Close Accordion” use_background_color_gradient=”off” background_color_gradient_start=”#2b87da” background_color_gradient_end=”#29c4a9″ background_color_gradient_type=”linear” background_color_gradient_direction=”180deg” background_color_gradient_direction_radial=”center” background_color_gradient_start_position=”0%” background_color_gradient_end_position=”100%” background_color_gradient_overlays_image=”off” parallax=”off” parallax_method=”on” background_size=”cover” background_position=”center” background_repeat=”no-repeat” background_blend=”normal” allow_player_pause=”off” background_video_pause_outside_viewport=”on” open_toggle_background_color=”#ffffff” closed_toggle_background_color=”#ffffff” text_shadow_style=”none” custom_padding=”0px|||” custom_padding_tablet=”0px|||” custom_padding_phone=”0px|||” box_shadow_style=”none” custom_css_main_element=”border:none;” custom_css_toggle_title=”font-size:0.8em;” custom_css_toggle_icon=”display:none;” /][/et_pb_accordion][/et_pb_column][/et_pb_row][/et_pb_section]
Post Disclaimer
Professional Scope of Practice *
The information on this blog site is not intended to replace a one-on-one relationship with a qualified healthcare professional or licensed physician and is not medical advice. We encourage you to make healthcare decisions based on your research and partnership with a qualified healthcare professional.
Blog Information & Scope Discussions
Welcome to El Paso's Premier Wellness and Injury Care Clinic & Wellness Blog, where Dr. Alex Jimenez, DC, FNP-C, a board-certified Family Practice Nurse Practitioner (FNP-BC) and Chiropractor (DC), presents insights on how our team is dedicated to holistic healing and personalized care. Our practice aligns with evidence-based treatment protocols inspired by integrative medicine principles, similar to those found on this site and our family practice-based chiromed.com site, focusing on restoring health naturally for patients of all ages.
Our areas of chiropractic practice include Wellness & Nutrition, Chronic Pain, Personal Injury, Auto Accident Care, Work Injuries, Back Injury, Low Back Pain, Neck Pain, Migraine Headaches, Sports Injuries, Severe Sciatica, Scoliosis, Complex Herniated Discs, Fibromyalgia, Chronic Pain, Complex Injuries, Stress Management, Functional Medicine Treatments, and in-scope care protocols.
Our information scope is limited to chiropractic, musculoskeletal, physical medicine, wellness, contributing etiological viscerosomatic disturbances within clinical presentations, associated somato-visceral reflex clinical dynamics, subluxation complexes, sensitive health issues, and functional medicine articles, topics, and discussions.
We provide and present clinical collaboration with specialists from various disciplines. Each specialist is governed by their professional scope of practice and their jurisdiction of licensure. We use functional health & wellness protocols to treat and support care for the injuries or disorders of the musculoskeletal system.
Our videos, posts, topics, subjects, and insights cover clinical matters and issues that relate to and directly or indirectly support our clinical scope of practice.*
Our office has made a reasonable effort to provide supportive citations and has identified relevant research studies that support our posts. We provide copies of supporting research studies available to regulatory boards and the public upon request.
We understand that we cover matters that require an additional explanation of how they may assist in a particular care plan or treatment protocol; therefore, to discuss the subject matter above further, please feel free to ask Dr. Alex Jimenez, DC, APRN, FNP-BC, or contact us at 915-850-0900.
We are here to help you and your family.
Blessings
Dr. Alex Jimenez DC, MSACP, APRN, FNP-BC*, CCST, IFMCP, CFMP, ATN
email: [email protected]
Licensed as a Doctor of Chiropractic (DC) in Texas & New Mexico*
Texas DC License # TX5807
New Mexico DC License # NM-DC2182
Licensed as a Registered Nurse (RN*) in Texas & Multistate
Texas RN License # 1191402
ANCC FNP-BC: Board Certified Nurse Practitioner*
Compact Status: Multi-State License: Authorized to Practice in 40 States*
Graduate with Honors: ICHS: MSN-FNP (Family Nurse Practitioner Program)
Degree Granted. Master's in Family Practice MSN Diploma (Cum Laude)
Dr. Alex Jimenez, DC, APRN, FNP-BC*, CFMP, IFMCP, ATN, CCST
(Board Certified: Family Practice Nurse Practitioner—Multistate)*
(Licensed Nurse Practitioner & Chiropractor - Multistate)*
Clinical Director
Digital Business Card
Dr. Maria Cardenas, MD
(Board Certified: Internal Medicine)
(Licensed Medical Doctor)
Medical Director, Clinical Director & Collaborative Physician
NPI # 1164426749
MD License #: J2933
Licenses and Board Certifications:
MD: Medical Doctor
DC: Doctor of Chiropractic
APRNP: Advanced Practice Registered Nurse
FNP-BC: Family Practice Specialization (Multi-State Board Certified)
RN: Registered Nurse (Multi-State Compact License)
CFMP: Certified Functional Medicine Provider
MSN-FNP: Master of Science in Family Practice Medicine
MSACP: Master of Science in Advanced Clinical Practice
IFMCP: Institute of Functional Medicine
CCST: Certified Chiropractic Spinal Trauma
ATN: Advanced Translational Neutrogenomics
Memberships & Associations:
TCA: Texas Chiropractic Association: Member ID: 104311
AANP: American Association of Nurse Practitioners: Member ID: 2198960
ANA: American Nurse Association: Member ID: 06458222 (District TX01)
TNA: Texas Nurse Association: Member ID: 06458222
NPI: 1205907805
| Primary Taxonomy | Selected Taxonomy | State | License Number |
|---|---|---|---|
| No | 111N00000X - Chiropractor | NM | DC2182 |
| Yes | 111N00000X - Chiropractor | TX | DC5807 |
| Yes | 363LF0000X - Nurse Practitioner - Family | TX | 1191402 |
| Yes | 363LF0000X - Nurse Practitioner - Family | FL | 11043890 |
| Yes | 363LF0000X - Nurse Practitioner - Family | CO | C-APN.0105610-C-NP |
| Yes | 363LF0000X - Nurse Practitioner - Family | NY | N25929 |
Dr. Alex Jimenez, DC, APRN, FNP-BC*, CFMP, IFMCP, ATN, CCST
(Board Certified: Family Practice Nurse Practitioner—Multistate)*
(Licensed Nurse Practitioner & Chiropractor - Multistate)*
Clinical Director
Digital Business Card
Dr. Maria Cardenas, MD
(Board Certified: Internal Medicine)*
(Licensed Medical Doctor)*
Medical Director, Clinical Director & Collaborative Physician
NPI # 1164426749
MD License #: J2933
Comments are closed.