

Functional Neurology: Chronic Excitotoxicity in Neurodegenerative Diseases

When compared to other central nervous system (CNS) health issues, chronic neurodegenerative diseases can be far more complicated. Foremostly, because the compromised mitochondrial function has been demonstrated in many neurodegenerative diseases, the resulting problems in energy sources are not as severe as the energy collapse in ischemic stroke. Therefore, if excitotoxicity contributes to neurodegeneration, a different time of chronic excitotoxicity needs to be assumed. In the following article, we will outline what is known about the pathways that may cause excitotoxicity in neurodegenerative diseases. We will specifically discuss that in amyotrophic lateral sclerosis (ALS), Alzheimer’s disease (AD) and Huntington’s disease (HD) as fundamental examples with sufficiently validated animal models in research studies.
Table of Contents
Amyotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease associated with the degeneration of motor neurons which ultimately determine the length of the health issue. ALS is considered fatal several years after it begins. It is hypothesized that L-glutamate excitotoxicity plays a role in the motor neuron death in ALS because cells demonstrate increased levels of calcium-permeable AMPA receptors and low levels of calcium-binding proteins. Compared to the utilization of AMPA and kainate, and L-HCA, in the spinal cord of rats, treatment with NMDA spared motor neurons suggests that NMDA excitotoxicity may actually not play a fundamental role in ALS. However, NMDA receptor-mediated excitotoxicity in motor neurons was demonstrated in chick embryo organotypic slice cultures. Electrophysiological research studies suggested that transient hyperexcitability of motor nerves in the presymptomatic phase of ALS in mice transgenic for the G93A mutation of human SOD1 is associated with hereditary ALS. Additionally, cortical hyperexcitability was recorded in familial and sporadic ALS patients with the onset of symptoms in familial ALS mutation carriers. Moreover, the only approved drug and/or medication utilized for ALS, which increases survival by 2 to 3 months, acts as an inhibitor of both NMDA and kainate receptors together with quickly upregulating EAAT activity in synaptosomes, according to several research studies.
In autopsied spinal cords from patients with ALS, several groups demonstrated a decrease in EAAT2 and not in EAAT1 protein expression in the gray matter of regions with considerable motor neuron loss. In addition, both L-glutamate uptake and EAAT2 immunoreactivity, as demonstrated by Western blotting, were demonstrated to be quantitatively decreased in postmortem tissue of ALS patients, particularly in the spinal cord, the tissue which is most commonly affected by the health issue. Additionally, it has been demonstrated that as a possible effect of EAAT2 downregulation, L-glutamate amounts are increased in the CSF in patients with ALS. However, this outcome measure couldn’t be replicated by other research studies.
The downregulation of EAAT2 in human ALS is demonstrated in several animal models of ALS, including transgenic mice expressing human SOD1 containing the G93A mutation which causes hereditary ALS or transgenic rats expressing the same mutation. Surprisingly, “whereas Bendotti demonstrated a late decrease in EAAT2 expression at the time when the mice had already become symptomatic,” research studies demonstrated fluctuations in EAAT2 expression at the presymptomatic stage. The β-lactam antibiotic ceftriaxone (Cef) promotes the production of EAAT2 in cultured murine spinal cord slices and in neuron/astrocyte co-cultures. In addition, it caused EAAT2 expression from the spinal cords of wild-type and mutant G93A mSOD1 Tg mice, which has been associated with a decrease in motor neuron loss, weight reduction, and other ALS-like symptoms as well as an increase in survival, compatible with the hypothesis that EAAT2 loss contributes to chronic excitotoxicity in this mouse model. Just recently, a significant decrease in EAAT2 immunoreactivity had been demonstrated in a separate bark model for ALS, rats expressing ALS-inducing mutant TAR DNA binding protein 43 in astrocytes only. Surprisingly, the research studies demonstrated that when measured by microdialysis, the extracellular L-glutamate and L-aspartate concentrations increase while the L-glutamate clearance capability decrease in the cerebral cortex of G93A mSOD1 Tg mice, however, this region doesn’t show overt pathology nor downregulation of EAAT1 when evaluated.
Taken together these research studies support the view that there is a downregulation of EAAT2 in both human ALS patients and animal models of ALS. However, while some animal research studies suggest that EAAT2 downregulation occurs before motor neuron loss, others are compatible with the hypothesis that the downregulation of EAAT2, the astroglial expression of which is associated with the existence of neurons, is a consequence of neurodegeneration in neurological diseases.
Furthermore, EAATs decrease extracellular L-glutamate, extracellular cerebral L-glutamate is upregulated in a variety of brain regions from the cystine/glutamate antiporter system x−c. XCT, one particular subunit of program x−c, was demonstrated to be differentially regulated and maintained in mouse models of ALS. Research studies demonstrated that the uptake of radiolabelled cystine was upregulated in spinal cord slices of presymptomatic G93A mSOD1 Tg mice at the age of 70 days but not in 55 or 100 days and not in symptomatic 130 day-old mice which also determined that the upregulation of cystine uptake at day 70 was because of system x−c activity utilizing the system x−c inhibitor sulfasalazine (SSZ). It needs to be considered, however, that cystine can also be hauled by EAATs. Therefore, as evidence about the SSZ-sensitivity of cystine uptakes were not demonstrated for days 100 and 130, the differential cystine uptake demonstrated in this research study at the older ages could rather be a result of decreased EAAT action. By comparison, research studies with rtPCR demonstrated a strong growth in xCT mRNA levels in G37R mSOD1 Tg mice on the beginning of symptoms, which has been further increased as symptoms improved. Moreover, it was demonstrated that xCT was primarily demonstrated in spinal cord microglial cells. Microglia revealed xCT mRNA upregulation in the presymptomatic stage. Taken together, these outcome measures suggest the system x−c is upregulated in animal models of ALS. However, the evidence is lacking about whether this is true for human cases of ALS. Nevertheless, further research studies revealed that the mRNA levels of CD68, a marker of microglial activation, were associated with xCT mRNA expression in postmortem spinal cord tissue of individuals with ALS, demonstrating that neuroinflammation in humans is also ultimately associated with xCT upregulation.
Beyond the dysregulation of L-glutamate and L-aspartate levels by EAAT downregulation or system x−c upregulation, pathways that indirectly regulate and maintain glutamatergic neurotransmission also have been suggested to participate in motor neuron degeneration in ALS. D-Serine levels have been shown to become considerably increased from the spinal cord of G93A mSOD1 Tg mice. Starting at disease onset and ongoing during the course of this symptomatic phase, D-serine increases NMDA excitotoxicity in motor neurons. The upregulation of D-serine at the spinal cord was duplicated by other research studies. Downregulation of this D-serine metabolizing enzyme DAO in the reticulospinal tract has been demonstrated as the main mechanism for D-serine upregulation in the spinal cord in ALS mice. In addition, genetic inactivation of DAO in mice has been associated with motor neuron degeneration and a deficiency in the D-serine generating enzyme serine racemase prolonged survival in G93A mSOD1 Tg mice although it hastened neurodegenerative disease onset. A heterozygous mutation of DAO has been demonstrated to be separate from the ALS phenotype in a large family with hereditary ALS. However, this continues to be the only family determined where a DAO mutation is associated with ALS.
Concerning the other amino acid co-agonist of the NMDA receptor, glycine, an increase in the CSF levels in patients with ALS was demonstrated by one group, however, it couldn’t be replicated by other research studies. Several research studies also determined that KYNA levels are upregulated in the CSF of bulbar ALS patients as well as those in end-stage ALS. Independently, it was revealed that tryptophan and KYN levels are increased in the CSF from ALS patients as compared to controls. Additionally, IDO was proven to be expressed in neurons and spinal cord microglia from patients with ALS, indicating that microglial activation may increase the conversion of tryptophan in ALS into KYN, among others.
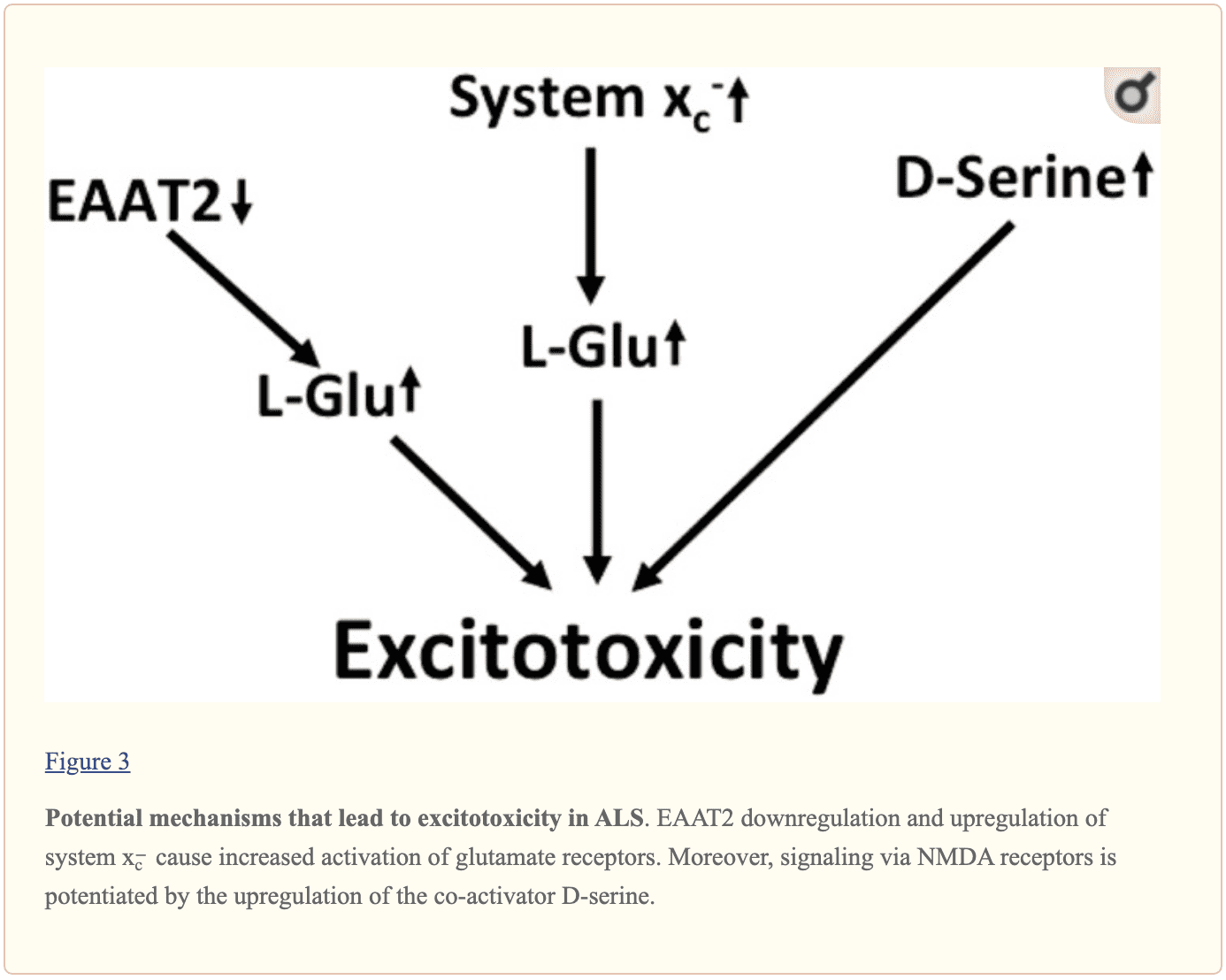
Multilayered evidence suggests that increased glutamatergic neurotransmission is within ALS and may ultimately cause neurodegeneration in neurodegenerative diseases, as shown in Figure 3. Downregulation of EAAT2 in astrocytes and upregulation of program x−forecast in the context of microglial activation was repeatedly documented. NMDA receptors by D-serine may also play a role in dysregulation. Moreover, the kynurenine pathway seems to be triggered in ALS.

In many research studies, evidence and outcome measures have demonstrated that chronic excitotoxicity may be associated with a variety of neurodegenerative diseases, including AD, HD, and ALS, ultimately causing neurodegeneration and a variery of symptoms associated with the health issues. The purpose of the following article is to outline what may cause excitotoxicity in neurodegenerative diseases. We will discuss these in amyotrophic lateral sclerosis (ALS), Alzheimer’s disease (AD) and Huntington’s disease (HD). – Dr. Alex Jimenez D.C., C.C.S.T. Insight – Dr. Alex Jimenez D.C., C.C.S.T. Insight
Metabolic Assessment Form
The following Metabolic Assessment Form can be filled out and presented to Dr. Alex Jimenez. Symptom groups listed on this form are not intended to be utilized as a diagnosis of any type of disease, condition, or any other type of health issue.
In the article above, we outlined what is known about the pathways which may cause excitotoxicity in neurodegenerative diseases. We also discussed that in amyotrophic lateral sclerosis (ALS), Alzheimer’s disease (AD) and Huntington’s disease (HD) as fundamental examples with sufficiently validated animal models in research studies. The scope of our information is limited to chiropractic, musculoskeletal and nervous health issues as well as functional medicine articles, topics, and discussions. We use functional health protocols to treat injuries or chronic disorders of the musculoskeletal system. To further discuss the subject matter above, please feel free to ask Dr. Alex Jimenez or contact us at 915-850-0900 .
Curated by Dr. Alex Jimenez
References
- Lewerenz, Jan, and Pamela Maher. “Chronic Glutamate Toxicity in Neurodegenerative Diseases-What Is the Evidence?” Frontiers in Neuroscience, Frontiers Media S.A., 16 Dec. 2015, www.ncbi.nlm.nih.gov/pmc/articles/PMC4679930/.
Additional Topic Discussion: Chronic Pain
Sudden pain is a natural response of the nervous system which helps to demonstrate possible injury. By way of instance, pain signals travel from an injured region through the nerves and spinal cord to the brain. Pain is generally less severe as the injury heals, however, chronic pain is different than the average type of pain. With chronic pain, the human body will continue sending pain signals to the brain, regardless if the injury has healed. Chronic pain can last for several weeks to even several years. Chronic pain can tremendously affect a patient’s mobility and it can reduce flexibility, strength, and endurance.
Neural Zoomer Plus for Neurological Disease

Dr. Alex Jimenez utilizes a series of tests to help evaluate neurological diseases. The Neural ZoomerTM Plus is an array of neurological autoantibodies which offers specific antibody-to-antigen recognition. The Vibrant Neural ZoomerTM Plus is designed to assess an individual’s reactivity to 48 neurological antigens with connections to a variety of neurologically related diseases. The Vibrant Neural ZoomerTM Plus aims to reduce neurological conditions by empowering patients and physicians with a vital resource for early risk detection and an enhanced focus on personalized primary prevention.
Formulas for Methylation Support

XYMOGEN’s Exclusive Professional Formulas are available through select licensed health care professionals. The internet sale and discounting of XYMOGEN formulas are strictly prohibited.
Proudly, Dr. Alexander Jimenez makes XYMOGEN formulas available only to patients under our care.
Please call our office in order for us to assign a doctor consultation for immediate access.
If you are a patient of Injury Medical & Chiropractic Clinic, you may inquire about XYMOGEN by calling 915-850-0900.
![]()
For your convenience and review of the XYMOGEN products please review the following link.*XYMOGEN-Catalog-Download
* All of the above XYMOGEN policies remain strictly in force.
Post Disclaimer
Professional Scope of Practice *
The information on this blog site is not intended to replace a one-on-one relationship with a qualified healthcare professional or licensed physician and is not medical advice. We encourage you to make healthcare decisions based on your research and partnership with a qualified healthcare professional.
Blog Information & Scope Discussions
Welcome to El Paso's Premier Wellness and Injury Care Clinic & Wellness Blog, where Dr. Alex Jimenez, DC, FNP-C, a board-certified Family Practice Nurse Practitioner (FNP-BC) and Chiropractor (DC), presents insights on how our team is dedicated to holistic healing and personalized care. Our practice aligns with evidence-based treatment protocols inspired by integrative medicine principles, similar to those found on this site and our family practice-based chiromed.com site, focusing on restoring health naturally for patients of all ages.
Our areas of chiropractic practice include Wellness & Nutrition, Chronic Pain, Personal Injury, Auto Accident Care, Work Injuries, Back Injury, Low Back Pain, Neck Pain, Migraine Headaches, Sports Injuries, Severe Sciatica, Scoliosis, Complex Herniated Discs, Fibromyalgia, Chronic Pain, Complex Injuries, Stress Management, Functional Medicine Treatments, and in-scope care protocols.
Our information scope is limited to chiropractic, musculoskeletal, physical medicine, wellness, contributing etiological viscerosomatic disturbances within clinical presentations, associated somato-visceral reflex clinical dynamics, subluxation complexes, sensitive health issues, and functional medicine articles, topics, and discussions.
We provide and present clinical collaboration with specialists from various disciplines. Each specialist is governed by their professional scope of practice and their jurisdiction of licensure. We use functional health & wellness protocols to treat and support care for the injuries or disorders of the musculoskeletal system.
Our videos, posts, topics, subjects, and insights cover clinical matters and issues that relate to and directly or indirectly support our clinical scope of practice.*
Our office has made a reasonable effort to provide supportive citations and has identified relevant research studies that support our posts. We provide copies of supporting research studies available to regulatory boards and the public upon request.
We understand that we cover matters that require an additional explanation of how they may assist in a particular care plan or treatment protocol; therefore, to discuss the subject matter above further, please feel free to ask Dr. Alex Jimenez, DC, APRN, FNP-BC, or contact us at 915-850-0900.
We are here to help you and your family.
Blessings
Dr. Alex Jimenez DC, MSACP, APRN, FNP-BC*, CCST, IFMCP, CFMP, ATN
email: [email protected]
Licensed as a Doctor of Chiropractic (DC) in Texas & New Mexico*
Texas DC License # TX5807
New Mexico DC License # NM-DC2182
Licensed as a Registered Nurse (RN*) in Texas & Multistate
Texas RN License # 1191402
ANCC FNP-BC: Board Certified Nurse Practitioner*
Compact Status: Multi-State License: Authorized to Practice in 40 States*
Graduate with Honors: ICHS: MSN-FNP (Family Nurse Practitioner Program)
Degree Granted. Master's in Family Practice MSN Diploma (Cum Laude)
Dr. Alex Jimenez, DC, APRN, FNP-BC*, CFMP, IFMCP, ATN, CCST
(Board Certified: Family Practice Nurse Practitioner—Multistate)*
(Licensed Nurse Practitioner & Chiropractor - Multistate)*
Clinical Director
Digital Business Card
Dr. Maria Cardenas, MD
(Board Certified: Internal Medicine)
(Licensed Medical Doctor)
Medical Director, Clinical Director & Collaborative Physician
NPI # 1164426749
MD License #: J2933
Licenses and Board Certifications:
MD: Medical Doctor
DC: Doctor of Chiropractic
APRNP: Advanced Practice Registered Nurse
FNP-BC: Family Practice Specialization (Multi-State Board Certified)
RN: Registered Nurse (Multi-State Compact License)
CFMP: Certified Functional Medicine Provider
MSN-FNP: Master of Science in Family Practice Medicine
MSACP: Master of Science in Advanced Clinical Practice
IFMCP: Institute of Functional Medicine
CCST: Certified Chiropractic Spinal Trauma
ATN: Advanced Translational Neutrogenomics
Memberships & Associations:
TCA: Texas Chiropractic Association: Member ID: 104311
AANP: American Association of Nurse Practitioners: Member ID: 2198960
ANA: American Nurse Association: Member ID: 06458222 (District TX01)
TNA: Texas Nurse Association: Member ID: 06458222
NPI: 1205907805
| Primary Taxonomy | Selected Taxonomy | State | License Number |
|---|---|---|---|
| No | 111N00000X - Chiropractor | NM | DC2182 |
| Yes | 111N00000X - Chiropractor | TX | DC5807 |
| Yes | 363LF0000X - Nurse Practitioner - Family | TX | 1191402 |
| Yes | 363LF0000X - Nurse Practitioner - Family | FL | 11043890 |
| Yes | 363LF0000X - Nurse Practitioner - Family | CO | C-APN.0105610-C-NP |
| Yes | 363LF0000X - Nurse Practitioner - Family | NY | N25929 |
Dr. Alex Jimenez, DC, APRN, FNP-BC*, CFMP, IFMCP, ATN, CCST
(Board Certified: Family Practice Nurse Practitioner—Multistate)*
(Licensed Nurse Practitioner & Chiropractor - Multistate)*
Clinical Director
Digital Business Card
Dr. Maria Cardenas, MD
(Board Certified: Internal Medicine)*
(Licensed Medical Doctor)*
Medical Director, Clinical Director & Collaborative Physician
NPI # 1164426749
MD License #: J2933
Comments are closed.